bitscore colors: <40, 40-50 , 50-80, 80-200, >200
BLASTP 2.2.30+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs", Nucleic Acids Res. 25:3389-3402.
Reference for composition-based statistics: Alejandro A. Schaffer,
L. Aravind, Thomas L. Madden, Sergei Shavirin, John L. Spouge, Yuri
I. Wolf, Eugene V. Koonin, and Stephen F. Altschul (2001),
"Improving the accuracy of PSI-BLAST protein database searches with
composition-based statistics and other refinements", Nucleic Acids
Res. 29:2994-3005.
Database: all_orgs
14,240,465 sequences; 5,121,972,263 total letters
Query= Contig-32_CDS_annotation_glimmer3.pl_2_2
Length=104
Score E
Sequences producing significant alignments: (Bits) Value
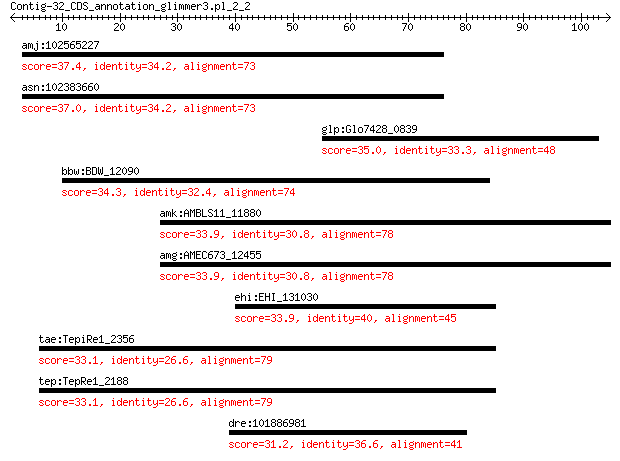
amj:102565227 PSD; pleckstrin and Sec7 domain containing 37.4 0.32
asn:102383660 PSD; pleckstrin and Sec7 domain containing 37.0 0.36
glp:Glo7428_0839 hypothetical protein 35.0 1.2
bbw:BDW_12090 hypothetical protein 34.3 3.1
amk:AMBLS11_11880 transporter (formate/nitrite transporter fam... 33.9 3.5
amg:AMEC673_12455 transporter (formate/nitrite transporter fam... 33.9 3.5
ehi:EHI_131030 27.t00024; hypothetical protein 33.9 4.2
tae:TepiRe1_2356 Transcriptional regulator, TrmB 33.1 6.9
tep:TepRe1_2188 transcriptional regulator TrmB 33.1 6.9
dre:101886981 homer protein homolog 1-like 31.2 8.3
> amj:102565227 PSD; pleckstrin and Sec7 domain containing
Length=788
Score = 37.4 bits (85), Expect = 0.32, Method: Compositional matrix adjust.
Identities = 25/73 (34%), Positives = 37/73 (51%), Gaps = 8/73 (11%)
Query 3 FSLFSPRVDYNRIHSAIKLSKEFFDVREKQSLQNLFSDSQSFESDWSDIFWNQTEERQRE 62
+P++ ++I ++KL KE F L +L DS + S +D W + EE QR
Sbjct 206 LPTLTPQIQ-DKIRDSVKLIKENF-----APLAHLEPDSGT--SSATDAPWTEREEEQRR 257
Query 63 FAESGLGSACRSE 75
A +GL S C SE
Sbjct 258 KANNGLHSPCHSE 270
> asn:102383660 PSD; pleckstrin and Sec7 domain containing
Length=858
Score = 37.0 bits (84), Expect = 0.36, Method: Compositional matrix adjust.
Identities = 25/73 (34%), Positives = 37/73 (51%), Gaps = 8/73 (11%)
Query 3 FSLFSPRVDYNRIHSAIKLSKEFFDVREKQSLQNLFSDSQSFESDWSDIFWNQTEERQRE 62
+P++ ++I ++KL KE F L +L DS + S +D W + EE QR
Sbjct 276 LPTLTPQIQ-DKIRDSVKLIKENF-----APLAHLEPDSGT--SSATDAPWTEREEEQRR 327
Query 63 FAESGLGSACRSE 75
A +GL S C SE
Sbjct 328 KANNGLHSPCHSE 340
> glp:Glo7428_0839 hypothetical protein
Length=269
Score = 35.0 bits (79), Expect = 1.2, Method: Compositional matrix adjust.
Identities = 16/48 (33%), Positives = 28/48 (58%), Gaps = 0/48 (0%)
Query 55 QTEERQREFAESGLGSACRSELSALVHSKVKHRHLNDMNNFFTERANY 102
+ E+R +E +SG+GS RS L ++ + + L +MN+ +R NY
Sbjct 219 EVEQRAQEQVQSGIGSGIRSLLLSIGYILIGWTGLRNMNSLPPDRPNY 266
> bbw:BDW_12090 hypothetical protein
Length=420
Score = 34.3 bits (77), Expect = 3.1, Method: Composition-based stats.
Identities = 24/75 (32%), Positives = 37/75 (49%), Gaps = 7/75 (9%)
Query 10 VDYNRIH-SAIKLSKEFFDVREKQSLQNLFSDSQSFESDWSDIFWNQTEERQREFAESGL 68
VDY + H A KL RE +SL+NL +++ SD + ++ ERQR +E G+
Sbjct 191 VDYRKAHIQANKLQ------REIESLENLIRNAKDDISDAKANYADEQRERQRSQSEGGI 244
Query 69 GSACRSELSALVHSK 83
C + S + K
Sbjct 245 CIDCMVQGSGYTYQK 259
> amk:AMBLS11_11880 transporter (formate/nitrite transporter family)
protein
Length=281
Score = 33.9 bits (76), Expect = 3.5, Method: Compositional matrix adjust.
Identities = 24/81 (30%), Positives = 34/81 (42%), Gaps = 11/81 (14%)
Query 27 DVREKQSLQNLFSDSQSFESDWSDIFWNQTEERQREFAE---SGLGSACRSELSALVHSK 83
DVR+ QSL ++ S ++ + EE QR F SG+ + LS L
Sbjct 24 DVRDNQSLNSV--------SLYAIVHREGLEELQRPFTSLWWSGVAAGIGISLSILAEGI 75
Query 84 VKHRHLNDMNNFFTERANYNV 104
+ H +N N F E Y V
Sbjct 76 LHHLFVNSPNQFVIENLGYTV 96
> amg:AMEC673_12455 transporter (formate/nitrite transporter family)
protein
Length=281
Score = 33.9 bits (76), Expect = 3.5, Method: Compositional matrix adjust.
Identities = 24/81 (30%), Positives = 34/81 (42%), Gaps = 11/81 (14%)
Query 27 DVREKQSLQNLFSDSQSFESDWSDIFWNQTEERQREFAE---SGLGSACRSELSALVHSK 83
DVR+ QSL ++ S ++ + EE QR F SG+ + LS L
Sbjct 24 DVRDNQSLNSV--------SLYAIVHREGLEELQRPFTSLWWSGVAAGIGISLSILAEGI 75
Query 84 VKHRHLNDMNNFFTERANYNV 104
+ H +N N F E Y V
Sbjct 76 LHHLFVNSPNQFVIENLGYTV 96
> ehi:EHI_131030 27.t00024; hypothetical protein
Length=1442
Score = 33.9 bits (76), Expect = 4.2, Method: Compositional matrix adjust.
Identities = 18/46 (39%), Positives = 25/46 (54%), Gaps = 1/46 (2%)
Query 40 DSQSFESDWSDIFWNQTEE-RQREFAESGLGSACRSELSALVHSKV 84
DSQ FE DIF E QR+F ++G S C SA++ +K+
Sbjct 285 DSQYFELHVKDIFIKSIYEILQRDFTDTGSSSECTKMFSAIIKNKL 330
> tae:TepiRe1_2356 Transcriptional regulator, TrmB
Length=900
Score = 33.1 bits (74), Expect = 6.9, Method: Composition-based stats.
Identities = 21/79 (27%), Positives = 42/79 (53%), Gaps = 2/79 (3%)
Query 6 FSPRVDYNRIHSAIKLSKEFFDVREKQSLQNLFSDSQSFESDWSDIFWNQTEERQREFAE 65
FSP D++ I S + F+DV E++ + + S E + S I ++ E+ +++ +
Sbjct 370 FSPDKDFHAIFSKDNSERHFYDVIEEK--LEILNKSGIEEKEISRILNSEIEKHFKKYIQ 427
Query 66 SGLGSACRSELSALVHSKV 84
+ +G + E+S +V KV
Sbjct 428 NMVGKYHKDEISKIVGEKV 446
> tep:TepRe1_2188 transcriptional regulator TrmB
Length=900
Score = 33.1 bits (74), Expect = 6.9, Method: Composition-based stats.
Identities = 21/79 (27%), Positives = 42/79 (53%), Gaps = 2/79 (3%)
Query 6 FSPRVDYNRIHSAIKLSKEFFDVREKQSLQNLFSDSQSFESDWSDIFWNQTEERQREFAE 65
FSP D++ I S + F+DV E++ + + S E + S I ++ E+ +++ +
Sbjct 370 FSPDKDFHAIFSKDNSERHFYDVIEEK--LEILNKSGIEEKEISRILNSEIEKHFKKYIQ 427
Query 66 SGLGSACRSELSALVHSKV 84
+ +G + E+S +V KV
Sbjct 428 NMVGKYHKDEISKIVGEKV 446
> dre:101886981 homer protein homolog 1-like
Length=67
Score = 31.2 bits (69), Expect = 8.3, Method: Compositional matrix adjust.
Identities = 15/41 (37%), Positives = 23/41 (56%), Gaps = 0/41 (0%)
Query 39 SDSQSFESDWSDIFWNQTEERQREFAESGLGSACRSELSAL 79
S + +S + N TEER RE ++ L + CR EL+A+
Sbjct 23 SAAGDLQSPVTPESINGTEERDRETPDAALNTGCRLELNAV 63
Lambda K H a alpha
0.319 0.130 0.382 0.792 4.96
Gapped
Lambda K H a alpha sigma
0.267 0.0410 0.140 1.90 42.6 43.6
Effective search space used: 126554967858