bitscore colors: <40, 40-50 , 50-80, 80-200, >200
BLASTP 2.2.24+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs", Nucleic Acids Res. 25:3389-3402.
Reference for composition-based statistics: Alejandro A. Schaffer,
L. Aravind, Thomas L. Madden, Sergei Shavirin, John L. Spouge, Yuri
I. Wolf, Eugene V. Koonin, and Stephen F. Altschul (2001),
"Improving the accuracy of PSI-BLAST protein database searches with
composition-based statistics and other refinements", Nucleic Acids
Res. 29:2994-3005.
Database: egene_temp_file_orthology_annotation_similarity_blast_database_966
164,496 sequences; 82,071,388 total letters
Query= Eace_1769_orf1
Length=165
Score E
Sequences producing significant alignments: (Bits) Value
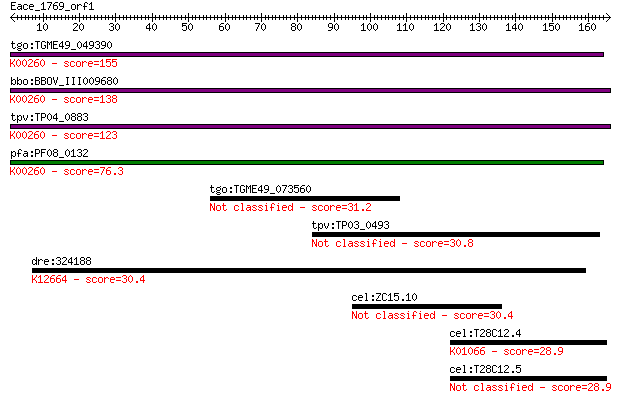
tgo:TGME49_049390 NAD-specific glutamate dehydrogenase, putati... 155 7e-38
bbo:BBOV_III009680 17.m07840; Glutamate/Leucine/Phenylalanine/... 138 1e-32
tpv:TP04_0883 NAD-specific glutamate dehydrogenase (EC:1.4.1.2... 123 3e-28
pfa:PF08_0132 glutamate dehydrogenase, putative (EC:1.4.1.2); ... 76.3 5e-14
tgo:TGME49_073560 kinesin heavy chain, putative 31.2 1.5
tpv:TP03_0493 hypothetical protein 30.8 1.9
dre:324188 cyp26b1, fc21d03, wu:fc21d03, wu:fc26h10, zgc:76999... 30.4 2.8
cel:ZC15.10 hypothetical protein 30.4 3.1
cel:T28C12.4 hypothetical protein; K01066 esterase / lipase [E... 28.9 8.2
cel:T28C12.5 hypothetical protein 28.9 9.0
> tgo:TGME49_049390 NAD-specific glutamate dehydrogenase, putative
(EC:1.4.1.2); K00260 glutamate dehydrogenase [EC:1.4.1.2]
Length=1113
Score = 155 bits (391), Expect = 7e-38, Method: Compositional matrix adjust.
Identities = 84/166 (50%), Positives = 118/166 (71%), Gaps = 7/166 (4%)
Query 1 YFRTAKPSSIADNLQAVIAARALHAVSGTEYFPEIQQIDERTGEVFALSKASLTAKRAAQ 60
YF+TA ++A NL VIAA+ L+ S T+YFP+IQQ ER GEVF L++ASL ++A+Q
Sbjct 136 YFQTATAQTVASNLACVIAAKILNEFSHTDYFPQIQQ--ERDGEVFLLARASLRNRKASQ 193
Query 61 NYKTERKLEQLFLNMGREENKQQMRMQCYRSTQSVFGIDAN-FERLRTYFFQKPDFPYR- 118
N++ ER++E+ FL G RMQCYRS+ S F A+ ERLRTYF Q+P++
Sbjct 194 NFQVEREIEKKFL--GLSGKTTPYRMQCYRSSGSFFDDPADENERLRTYFLQQPEYCAET 251
Query 119 -EEELSPTEKDISKLLDVNFFASKKNTVTASIFQRLNEQLVGDSTG 163
++ + P+E D+SKLLDV FFA+K+NT TA I++RLNE++V DS+G
Sbjct 252 TKDGVDPSETDLSKLLDVYFFANKRNTATAEIYRRLNEEVVRDSSG 297
> bbo:BBOV_III009680 17.m07840; Glutamate/Leucine/Phenylalanine/Valine
dehydrogenase family protein (EC:1.4.1.2); K00260 glutamate
dehydrogenase [EC:1.4.1.2]
Length=1025
Score = 138 bits (347), Expect = 1e-32, Method: Compositional matrix adjust.
Identities = 73/166 (43%), Positives = 107/166 (64%), Gaps = 6/166 (3%)
Query 1 YFRTAKPSSIADNLQAVIAARALHAVSGTEYFPEIQQIDERTGEVFALSKASLTAKRAAQ 60
YF T+ IA+N+ +V+ A+ LH SG++YFP I+Q+ + G VF +++ASL + +Q
Sbjct 73 YFSTSNAKMIANNMVSVLTAKILHENSGSDYFPLIEQVHD--GRVFIITRASLLNHKLSQ 130
Query 61 NYKTERKLEQLFLNMGREENKQQMRMQCYRSTQSVFGIDAN-FERLRTYFFQKPDFPYRE 119
Y E+ +EQ FL+ G + + RMQC+RST S+F AN FERLR YFFQ P+F E
Sbjct 131 IYAVEKTVEQRFLHFG-DVTRPVWRMQCFRSTNSMFDDPANLFERLRIYFFQLPNF--AE 187
Query 120 EELSPTEKDISKLLDVNFFASKKNTVTASIFQRLNEQLVGDSTGLA 165
P E +SKLLD +F+ +K+NT+T IF +LN+Q+V G+
Sbjct 188 NNPEPGELRLSKLLDKDFYKNKRNTITEDIFYKLNKQIVMSDGGIG 233
> tpv:TP04_0883 NAD-specific glutamate dehydrogenase (EC:1.4.1.2);
K00260 glutamate dehydrogenase [EC:1.4.1.2]
Length=1178
Score = 123 bits (309), Expect = 3e-28, Method: Compositional matrix adjust.
Identities = 64/166 (38%), Positives = 106/166 (63%), Gaps = 6/166 (3%)
Query 1 YFRTAKPSSIADNLQAVIAARALHAVSGTEYFPEIQQIDERTGEVFALSKASLTAKRAAQ 60
YF ++ + IA+N+ +V++A+ LH SG+ +FP I+QI E + F +++ S +++++
Sbjct 72 YFSSSPTALIANNVISVMSAKVLHENSGSNFFPTIEQITEDSA--FIIARTSSVNRKSSE 129
Query 61 NYKTERKLEQLFLNMGREENKQQMRMQCYRSTQSVFG-IDANFERLRTYFFQKPDFPYRE 119
NY E+++E + N+ ++ K RMQC+RS+ SVF + F+RL+TYF QKP F
Sbjct 130 NYLVEQRVEGKYFNLD-DDRKTCWRMQCFRSSGSVFKETEHIFDRLKTYFLQKPKFTVNN 188
Query 120 EELSPTEKDISKLLDVNFFASKKNTVTASIFQRLNEQLVGDSTGLA 165
+ P E +++ LLD F+ +KKNT+T IF LN++LV TGL
Sbjct 189 PD--PGETELANLLDREFYLNKKNTITEQIFHNLNKKLVESKTGLG 232
> pfa:PF08_0132 glutamate dehydrogenase, putative (EC:1.4.1.2);
K00260 glutamate dehydrogenase [EC:1.4.1.2]
Length=1397
Score = 76.3 bits (186), Expect = 5e-14, Method: Composition-based stats.
Identities = 47/164 (28%), Positives = 84/164 (51%), Gaps = 6/164 (3%)
Query 1 YFRTAKPSSIADNLQAVIAARALHAVSGTEYFPEIQQIDERTGEVFALSKASLTAKRAAQ 60
+F P I+ + +I A+ S +YFP ++ + +F +++ +
Sbjct 71 HFEETSPELISKVVVCIITAKINEQYSSDKYFPTFEETHDNV--IFIITRVFADDNKTRL 128
Query 61 NYKTERKLEQLFLNMGREENKQQMRMQCYRSTQSVFGIDANF-ERLRTYFFQKPDFPYRE 119
NYK E+K+E+ + N + +K R++ +RS SVF + + E LRTY + P Y +
Sbjct 129 NYKMEKKIEEKYFNFS-DMSKDCYRLKSFRSVHSVFDKEHTYQEPLRTYILELPT--YND 185
Query 120 EELSPTEKDISKLLDVNFFASKKNTVTASIFQRLNEQLVGDSTG 163
+ + E D+ KL+DVNF+ K T + I+ LN+ ++ D TG
Sbjct 186 DIIKENETDLKKLMDVNFYNYIKGTRSEQIYYELNKAVLYDLTG 229
> tgo:TGME49_073560 kinesin heavy chain, putative
Length=1641
Score = 31.2 bits (69), Expect = 1.5, Method: Compositional matrix adjust.
Identities = 16/53 (30%), Positives = 32/53 (60%), Gaps = 1/53 (1%)
Query 56 KRAAQNYKTERKLEQLFLNMGREENK-QQMRMQCYRSTQSVFGIDANFERLRT 107
+RAAQ+ + +++QL +++ ++ NK QQ M+ + T VFG+ + +T
Sbjct 484 QRAAQSPENNEEVQQLKIDLEKKINKFQQFIMKATKPTAGVFGVSGTLSKRKT 536
> tpv:TP03_0493 hypothetical protein
Length=624
Score = 30.8 bits (68), Expect = 1.9, Method: Composition-based stats.
Identities = 26/100 (26%), Positives = 41/100 (41%), Gaps = 21/100 (21%)
Query 84 MRMQC---YRSTQSVFGIDA-----------NFERLRTYFFQKPDFPYREEELSPTEKDI 129
+RM C R +FG+D NFE + ++ + DF R+ L T+K +
Sbjct 215 LRMPCKHVLRYVSWLFGVDPCHVLIFSGNPQNFETIGSHLYSLDDFSNRDNSLGYTQKPL 274
Query 130 SKLLDV-------NFFASKKNTVTASIFQRLNEQLVGDST 162
L + N N +A + QRL++ L D T
Sbjct 275 IHLFTIQGNLVMPNNLNLYHNPASARLSQRLSQSLSYDPT 314
> dre:324188 cyp26b1, fc21d03, wu:fc21d03, wu:fc26h10, zgc:76999;
cytochrome P450, family 26, subfamily b, polypeptide 1;
K12664 cytochrome P450, family 26, subfamily B
Length=511
Score = 30.4 bits (67), Expect = 2.8, Method: Composition-based stats.
Identities = 42/172 (24%), Positives = 70/172 (40%), Gaps = 27/172 (15%)
Query 7 PSSIADNLQAV------IAARALHAVSGTEYFPEIQQIDERTGEVFALSKASLTAKRAAQ 60
P+S+A+++ + I A+ + Y P+IQQ+ + T V++ + + R +Q
Sbjct 127 PNSLANSIGDIHRKRRKIFAKVFSHEALESYLPKIQQVIQETLRVWSSNPDPINVYRESQ 186
Query 61 NYKTERKLEQLF-LNMGREENKQQMRMQCYRST-----QSVFG--IDANFE------RLR 106
+ L + EE M C ST ++VF ID F R R
Sbjct 187 RLSFNMAVRVLLGFRIPEEE------MHCLFSTFQEFVENVFSLPIDLPFSGYRKGIRAR 240
Query 107 TYFFQKPDFPYREEELSPTEKDISKLLDVNFFASKKNTVTASIFQRLNEQLV 158
+ + RE+ L KD + LDV ++K+N T Q L E +
Sbjct 241 DSLQKSIEKAIREKPLHTQGKDYTDALDVLLESAKENN-TELTMQELKESTI 291
> cel:ZC15.10 hypothetical protein
Length=196
Score = 30.4 bits (67), Expect = 3.1, Method: Compositional matrix adjust.
Identities = 16/41 (39%), Positives = 23/41 (56%), Gaps = 2/41 (4%)
Query 95 VFGIDANFERLRTYFFQKPDFPYREEELSPTEKDISKLLDV 135
V+G NF R R YF + D P+ E+ +S T I LL++
Sbjct 105 VYGAVLNFHRNRAYFVK--DMPFVEDLISDTATTIRPLLEL 143
> cel:T28C12.4 hypothetical protein; K01066 esterase / lipase
[EC:3.1.1.-]
Length=658
Score = 28.9 bits (63), Expect = 8.2, Method: Composition-based stats.
Identities = 16/43 (37%), Positives = 26/43 (60%), Gaps = 3/43 (6%)
Query 122 LSPTEKDISKLLDVNFFASKKNTVTASIFQRLNEQLVGDSTGL 164
L TEK+I+ +DV S+KN ++ S + + E++ GDS L
Sbjct 428 LRGTEKEINSAIDV---LSRKNRLSRSKIEAMTEKVYGDSPAL 467
> cel:T28C12.5 hypothetical protein
Length=539
Score = 28.9 bits (63), Expect = 9.0, Method: Composition-based stats.
Identities = 16/43 (37%), Positives = 25/43 (58%), Gaps = 3/43 (6%)
Query 122 LSPTEKDISKLLDVNFFASKKNTVTASIFQRLNEQLVGDSTGL 164
L TEK+I +DV S+KN ++ S + + E++ GDS L
Sbjct 321 LRGTEKEIDSAIDV---LSRKNGLSRSKIETMTEKVYGDSPAL 360
Lambda K H
0.317 0.130 0.355
Gapped
Lambda K H
0.267 0.0410 0.140
Effective search space used: 3962792044
Database: egene_temp_file_orthology_annotation_similarity_blast_database_966
Posted date: Sep 16, 2011 8:45 PM
Number of letters in database: 82,071,388
Number of sequences in database: 164,496
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Neighboring words threshold: 11
Window for multiple hits: 40