bitscore colors: <40, 40-50 , 50-80, 80-200, >200
BLASTP 2.2.24+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs", Nucleic Acids Res. 25:3389-3402.
Reference for composition-based statistics: Alejandro A. Schaffer,
L. Aravind, Thomas L. Madden, Sergei Shavirin, John L. Spouge, Yuri
I. Wolf, Eugene V. Koonin, and Stephen F. Altschul (2001),
"Improving the accuracy of PSI-BLAST protein database searches with
composition-based statistics and other refinements", Nucleic Acids
Res. 29:2994-3005.
Database: egene_temp_file_orthology_annotation_similarity_blast_database_865
164,496 sequences; 82,071,388 total letters
Query= Emax_0194_orf1
Length=128
Score E
Sequences producing significant alignments: (Bits) Value
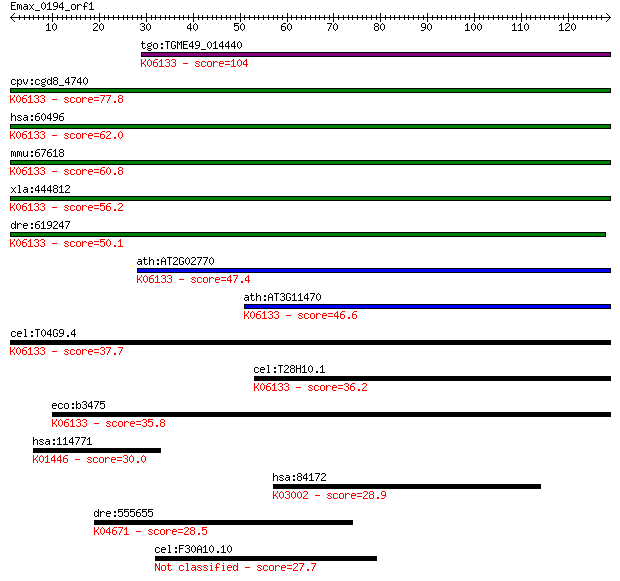
tgo:TGME49_014440 4'-phosphopantetheinyl transferase, putative... 104 8e-23
cpv:cgd8_4740 phosphopantetheinyl transferase ; K06133 4'-phos... 77.8 7e-15
hsa:60496 AASDHPPT, AASD-PPT, DKFZp566E2346, LYS2, LYS5; amino... 62.0 5e-10
mmu:67618 Aasdhppt, 2010309J24Rik, 2810407B07Rik, AASD-PPT, CG... 60.8 1e-09
xla:444812 aasdhppt, MGC84206; aminoadipate-semialdehyde dehyd... 56.2 2e-08
dre:619247 MGC114148; zgc:114148; K06133 4'-phosphopantetheiny... 50.1 2e-06
ath:AT2G02770 holo-[acyl-carrier-protein] synthase/ magnesium ... 47.4 1e-05
ath:AT3G11470 4'-phosphopantetheinyl transferase family protei... 46.6 2e-05
cel:T04G9.4 hypothetical protein; K06133 4'-phosphopantetheiny... 37.7 0.009
cel:T28H10.1 hypothetical protein; K06133 4'-phosphopantethein... 36.2 0.024
eco:b3475 acpT, ECK3459, JW3440, yhhU; holo-(acyl carrier prot... 35.8 0.031
hsa:114771 PGLYRP3, MGC149197, PGRP-Ialpha, PGRPIA; peptidogly... 30.0 1.9
hsa:84172 POLR1B, FLJ10816, FLJ21921, MGC131780, RPA135, RPA2,... 28.9 4.3
dre:555655 bmpr2b; bone morphogenetic protein receptor, type I... 28.5 5.1
cel:F30A10.10 hypothetical protein 27.7 8.2
> tgo:TGME49_014440 4'-phosphopantetheinyl transferase, putative
(EC:2.7.8.7); K06133 4'-phosphopantetheinyl transferase [EC:2.7.8.-]
Length=437
Score = 104 bits (259), Expect = 8e-23, Method: Compositional matrix adjust.
Identities = 52/106 (49%), Positives = 70/106 (66%), Gaps = 6/106 (5%)
Query 29 IERKETRSKPIWKPSPGETADFVHFNVSHDEGLVVFGASRHLVGVDCMRCSPRG---RDI 85
IER+ + KPIW P E + F+HFNVSHD GLVV ASR LVGVDCM+ + RG R +
Sbjct 123 IERQRSGQKPIWIPHAEERSPFIHFNVSHDTGLVVLSASRLLVGVDCMQVTVRGASSRSV 182
Query 86 SSFLQQMRRHCTLRDWSYIIAVH---TPEQQLRRFMRVWTVKEAFV 128
+ L+ M C R+ Y++ + E++LRRFM++WT+KEAFV
Sbjct 183 ADLLRAMDSQCVGRERPYVLGEDSDVSDEEKLRRFMKIWTMKEAFV 228
> cpv:cgd8_4740 phosphopantetheinyl transferase ; K06133 4'-phosphopantetheinyl
transferase [EC:2.7.8.-]
Length=267
Score = 77.8 bits (190), Expect = 7e-15, Method: Compositional matrix adjust.
Identities = 49/138 (35%), Positives = 74/138 (53%), Gaps = 18/138 (13%)
Query 1 DRLRALSSRLLQRLLLSSYCCCNPQHIVIERKETRSKPIWKPSPGETADFVHFNVSHDEG 60
DR R+L S LL +L L +Y +P+ + I R E KP +K E+ +HFNVSHD
Sbjct 8 DRKRSLLSVLLMKLALMNYYDISPKQVKIIR-EKGMKPYFK-YDSESDSLLHFNVSHDGD 65
Query 61 LVVFGASRHLVGVDCMRC--SPRGRDISS--------FLQQMRRHCTLRDWSYIIAVHTP 110
+VV S+++VG+D M+ SP +S+ FL M+ +W YI
Sbjct 66 VVVIILSKYMVGIDIMKTELSPSRVQLSNNIMEANEKFLNNMKNVFHPSEWEYI------ 119
Query 111 EQQLRRFMRVWTVKEAFV 128
++ + +FM WT+KE+FV
Sbjct 120 QKDISKFMHYWTIKESFV 137
> hsa:60496 AASDHPPT, AASD-PPT, DKFZp566E2346, LYS2, LYS5; aminoadipate-semialdehyde
dehydrogenase-phosphopantetheinyl transferase
(EC:2.7.8.-); K06133 4'-phosphopantetheinyl transferase
[EC:2.7.8.-]
Length=309
Score = 62.0 bits (149), Expect = 5e-10, Method: Compositional matrix adjust.
Identities = 40/130 (30%), Positives = 60/130 (46%), Gaps = 3/130 (2%)
Query 1 DRLRALSSRLLQRLLLSSYCCCNPQHIVIERKETRSKPIWKPSPGETADFVHFNVSHDEG 60
D A++ RL+ R L++ HI ++R + KP+ +FN+SH
Sbjct 56 DAKAAMAGRLMIRKLVAEKLNIPWNHIRLQRT-AKGKPVLAKDSSNPYPNFNFNISHQGD 114
Query 61 LVVFGASRHL-VGVDCMRCSPRGR-DISSFLQQMRRHCTLRDWSYIIAVHTPEQQLRRFM 118
V A L VG+D M+ S GR I F M+R T ++W I + QL F
Sbjct 115 YAVLAAEPELQVGIDIMKTSFPGRGSIPEFFHIMKRKFTNKEWETIRSFKDEWTQLDMFY 174
Query 119 RVWTVKEAFV 128
R W +KE+F+
Sbjct 175 RNWALKESFI 184
> mmu:67618 Aasdhppt, 2010309J24Rik, 2810407B07Rik, AASD-PPT,
CGI-80, LYS2, LYS5; aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyl
transferase (EC:2.7.8.-); K06133 4'-phosphopantetheinyl
transferase [EC:2.7.8.-]
Length=309
Score = 60.8 bits (146), Expect = 1e-09, Method: Compositional matrix adjust.
Identities = 40/130 (30%), Positives = 62/130 (47%), Gaps = 3/130 (2%)
Query 1 DRLRALSSRLLQRLLLSSYCCCNPQHIVIERKETRSKPIWKPSPGETADFVHFNVSHDEG 60
D AL+ RL+ R L++ HI ++R ++ KP+ +FN+SH
Sbjct 56 DAKAALAGRLMIRKLVAEKLNIPWDHIRLQRT-SKGKPVLAKDSLNPYPNFNFNISHQGD 114
Query 61 LVVFGASRHL-VGVDCMRCSPRGR-DISSFLQQMRRHCTLRDWSYIIAVHTPEQQLRRFM 118
V A + VG+D M+ S GR I F M+R T ++W I + + QL F
Sbjct 115 YAVLAAEPEVQVGIDIMKTSFPGRGSIPEFFHIMKRKFTKKEWETIRSFNDEWTQLDMFY 174
Query 119 RVWTVKEAFV 128
R W +KE+F+
Sbjct 175 RHWALKESFI 184
> xla:444812 aasdhppt, MGC84206; aminoadipate-semialdehyde dehydrogenase-phosphopantetheinyl
transferase; K06133 4'-phosphopantetheinyl
transferase [EC:2.7.8.-]
Length=302
Score = 56.2 bits (134), Expect = 2e-08, Method: Compositional matrix adjust.
Identities = 38/130 (29%), Positives = 59/130 (45%), Gaps = 3/130 (2%)
Query 1 DRLRALSSRLLQRLLLSSYCCCNPQHIVIERKETRSKPIWKPSPGETADFVHFNVSHDEG 60
D A++ RLL R +++ I++ER + KP +FNVSH
Sbjct 53 DAKAAMAGRLLMRKVIADKLQIPWDRILLERT-GKGKPFLTGGSSSEYPCFNFNVSHQGD 111
Query 61 LVVFGAS-RHLVGVDCMRCS-PRGRDISSFLQQMRRHCTLRDWSYIIAVHTPEQQLRRFM 118
V A VGVD M+ P I F + M R T ++W+ I +++ +L F
Sbjct 112 YAVLAAEPDRQVGVDIMKTDLPGSSSIEEFFRLMNRQFTEKEWNSIRSMNNDWARLDMFY 171
Query 119 RVWTVKEAFV 128
R W +KE+F+
Sbjct 172 RHWALKESFI 181
> dre:619247 MGC114148; zgc:114148; K06133 4'-phosphopantetheinyl
transferase [EC:2.7.8.-]
Length=293
Score = 50.1 bits (118), Expect = 2e-06, Method: Compositional matrix adjust.
Identities = 40/129 (31%), Positives = 57/129 (44%), Gaps = 5/129 (3%)
Query 1 DRLRALSSRLLQRLLLSSYCCCNPQHIVIERKETRSKPIWKPSPGETADFVHFNVSHDEG 60
D A++ RLL R L+ ++R E R KP + +A FNVSH
Sbjct 43 DAKSAMAGRLLIRKLVCEKMGFAWDGFRLQRTE-RGKPYLPQT--SSAPSWSFNVSHQGD 99
Query 61 LVVFGA-SRHLVGVDCMRCS-PRGRDISSFLQQMRRHCTLRDWSYIIAVHTPEQQLRRFM 118
V A + VG+D M+ S P + F + M R T +W+ I + QL F
Sbjct 100 YAVLAAEAGRQVGIDVMKTSRPGSSSVQEFFRIMNRQFTDLEWTNIRTAGSDWDQLHMFY 159
Query 119 RVWTVKEAF 127
R W +KE+F
Sbjct 160 RHWALKESF 168
> ath:AT2G02770 holo-[acyl-carrier-protein] synthase/ magnesium
ion binding; K06133 4'-phosphopantetheinyl transferase [EC:2.7.8.-]
Length=661
Score = 47.4 bits (111), Expect = 1e-05, Method: Composition-based stats.
Identities = 29/104 (27%), Positives = 53/104 (50%), Gaps = 5/104 (4%)
Query 28 VIERKETRSKP--IWKPSPGETADFVHFNVSHDEGLVVFGASRHL-VGVDCMRCSPRGRD 84
+I +K KP W+ + +HFN+SH + L+ G + H+ VG+D + +
Sbjct 404 LIFKKNMYGKPEVDWQSYKNCDSPPLHFNISHTDSLISCGVTVHVPVGIDLEEMERKIK- 462
Query 85 ISSFLQQMRRHCTLRDWSYIIAVHTPEQQLRRFMRVWTVKEAFV 128
L R + + ++ A+ PE Q + F+++WT+KEA+V
Sbjct 463 -HDVLALAERFYSADEVKFLSAIPDPEVQRKEFIKLWTLKEAYV 505
> ath:AT3G11470 4'-phosphopantetheinyl transferase family protein;
K06133 4'-phosphopantetheinyl transferase [EC:2.7.8.-]
Length=231
Score = 46.6 bits (109), Expect = 2e-05, Method: Compositional matrix adjust.
Identities = 25/80 (31%), Positives = 47/80 (58%), Gaps = 5/80 (6%)
Query 51 VHFNVSHDEGLVVFGASRHL-VGVDCMRCSPRGR-DISSFLQQMRRHCTLRDWSYIIAVH 108
+HFN+SH + L+ G + H+ VG+D + + DI +F + R + + ++ +
Sbjct 55 LHFNISHTDSLIACGVTVHVPVGIDVEDKERKIKHDILAFAE---RFYSADEVKFLSTLP 111
Query 109 TPEQQLRRFMRVWTVKEAFV 128
PE Q + F+++WT+KEA+V
Sbjct 112 DPEVQRKEFIKLWTLKEAYV 131
> cel:T04G9.4 hypothetical protein; K06133 4'-phosphopantetheinyl
transferase [EC:2.7.8.-]
Length=297
Score = 37.7 bits (86), Expect = 0.009, Method: Compositional matrix adjust.
Identities = 37/131 (28%), Positives = 52/131 (39%), Gaps = 7/131 (5%)
Query 1 DRLRALSSRLLQRLLLSSYCCCNPQHIVIERKETRSKPIWKPSPGETADFVHFNVSHDEG 60
D L L RLL R + I ER E R KP P +T NVSH
Sbjct 52 DALACLFGRLLLRHSAQKFSGEPWNTIRFERTE-RGKPFL-AVPADTT--FGLNVSHQGD 107
Query 61 LVVFGAS-RHLVGVDCMRCSPR--GRDISSFLQQMRRHCTLRDWSYIIAVHTPEQQLRRF 117
V F +S VGVD MR + ++ M + + + + + T ++ F
Sbjct 108 YVAFASSCSSKVGVDVMRLDNERNNKTADEYINSMAKSASPEELRMMRSQPTEAMKMTMF 167
Query 118 MRVWTVKEAFV 128
R W +KEA +
Sbjct 168 YRYWCLKEAIL 178
> cel:T28H10.1 hypothetical protein; K06133 4'-phosphopantetheinyl
transferase [EC:2.7.8.-]
Length=299
Score = 36.2 bits (82), Expect = 0.024, Method: Compositional matrix adjust.
Identities = 24/80 (30%), Positives = 40/80 (50%), Gaps = 6/80 (7%)
Query 53 FNVSHDEGLVVFGASRHLVGVDCMRCSPRGRDISSF-LQQMRRHCTLRDWSYIIAVHTPE 111
+NVSH LVV +GVD MR + R+ +S + ++RH + + + E
Sbjct 97 YNVSHHGDLVVLATGDTRIGVDVMRVNEARRETASEQMNTLKRHFSENEIEMVKGGDKCE 156
Query 112 QQLRR---FMRVWTVKEAFV 128
L+R F R+W +KE+ +
Sbjct 157 --LKRWHAFYRIWCLKESIL 174
> eco:b3475 acpT, ECK3459, JW3440, yhhU; holo-(acyl carrier protein)
synthase 2 (EC:2.7.8.7); K06133 4'-phosphopantetheinyl
transferase [EC:2.7.8.-]
Length=195
Score = 35.8 bits (81), Expect = 0.031, Method: Compositional matrix adjust.
Identities = 37/122 (30%), Positives = 54/122 (44%), Gaps = 19/122 (15%)
Query 10 LLQRLLLSSYCCCNPQHIVIERKETRSKPIWKPSPGETADFVHFNVSHD-EGLVVFGASR 68
L R LLS P+ I E+ KP + P + FN+SH + + + +
Sbjct 35 LAGRALLSHTLSPLPEIIYGEQ----GKPAFAPEMP-----LWFNLSHSGDDIALLLSDE 85
Query 69 HLVGVDCMRCSPRG--RDISSFLQQMRRHCTLRDWSYIIAVHTPEQQLRRFMRVWTVKEA 126
VG D PR R +++ + + H + AVH P+QQL F R+WT KEA
Sbjct 86 GEVGCDIEVIRPRANWRWLANAVFSLGEHAEMD------AVH-PDQQLEMFWRIWTRKEA 138
Query 127 FV 128
V
Sbjct 139 IV 140
> hsa:114771 PGLYRP3, MGC149197, PGRP-Ialpha, PGRPIA; peptidoglycan
recognition protein 3; K01446 N-acetylmuramoyl-L-alanine
amidase [EC:3.5.1.28]
Length=341
Score = 30.0 bits (66), Expect = 1.9, Method: Compositional matrix adjust.
Identities = 14/27 (51%), Positives = 17/27 (62%), Gaps = 0/27 (0%)
Query 6 LSSRLLQRLLLSSYCCCNPQHIVIERK 32
LS R +Q LLL C +PQH V+ RK
Sbjct 150 LSPRYIQPLLLKEETCLDPQHPVMPRK 176
> hsa:84172 POLR1B, FLJ10816, FLJ21921, MGC131780, RPA135, RPA2,
Rpo1-2; polymerase (RNA) I polypeptide B, 128kDa (EC:2.7.7.6);
K03002 DNA-directed RNA polymerase I subunit RPA2 [EC:2.7.7.6]
Length=1079
Score = 28.9 bits (63), Expect = 4.3, Method: Composition-based stats.
Identities = 15/58 (25%), Positives = 23/58 (39%), Gaps = 1/58 (1%)
Query 57 HDEGLVVFGASRHLVG-VDCMRCSPRGRDISSFLQQMRRHCTLRDWSYIIAVHTPEQQ 113
D GL V + + + RC RG D + R W ++ VHTP+ +
Sbjct 393 QDSGLCVVADKLNFIRYLSHFRCVHRGADFAKMRTTTVRRLLPESWGFLCPVHTPDGE 450
> dre:555655 bmpr2b; bone morphogenetic protein receptor, type
II b (serine/threonine kinase) (EC:2.7.11.30); K04671 bone
morphogenetic protein receptor type-2 [EC:2.7.11.30]
Length=1071
Score = 28.5 bits (62), Expect = 5.1, Method: Composition-based stats.
Identities = 16/55 (29%), Positives = 24/55 (43%), Gaps = 5/55 (9%)
Query 19 YCCCNPQHIVIERKETRSKPIWKPSPGETADFVHFNVSHDEGLVVFGASRHLVGV 73
+CCC+ + E W PSP TA+ + DE + + AS +V V
Sbjct 122 FCCCSTNMCNVNFTEN-----WVPSPTSTANRDQPPIHRDEAIFIALASVSIVAV 171
> cel:F30A10.10 hypothetical protein
Length=1262
Score = 27.7 bits (60), Expect = 8.2, Method: Composition-based stats.
Identities = 15/50 (30%), Positives = 26/50 (52%), Gaps = 3/50 (6%)
Query 32 KETRSKPIWKPSPGETA---DFVHFNVSHDEGLVVFGASRHLVGVDCMRC 78
KET KP +P+ + A + ++ V+ DE V F + + +DC +C
Sbjct 3 KETDKKPKERPNQRKVAFRNEAINALVNTDEDQVTFSKAMEVAKLDCQKC 52
Lambda K H
0.328 0.138 0.452
Gapped
Lambda K H
0.267 0.0410 0.140
Effective search space used: 2049573556
Database: egene_temp_file_orthology_annotation_similarity_blast_database_865
Posted date: Sep 17, 2011 11:19 AM
Number of letters in database: 82,071,388
Number of sequences in database: 164,496
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Neighboring words threshold: 11
Window for multiple hits: 40