bitscore colors: <40, 40-50 , 50-80, 80-200, >200
BLASTP 2.2.24+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs", Nucleic Acids Res. 25:3389-3402.
Reference for composition-based statistics: Alejandro A. Schaffer,
L. Aravind, Thomas L. Madden, Sergei Shavirin, John L. Spouge, Yuri
I. Wolf, Eugene V. Koonin, and Stephen F. Altschul (2001),
"Improving the accuracy of PSI-BLAST protein database searches with
composition-based statistics and other refinements", Nucleic Acids
Res. 29:2994-3005.
Database: egene_temp_file_orthology_annotation_similarity_blast_database_866
164,496 sequences; 82,071,388 total letters
Query= Eten_0885_orf1
Length=129
Score E
Sequences producing significant alignments: (Bits) Value
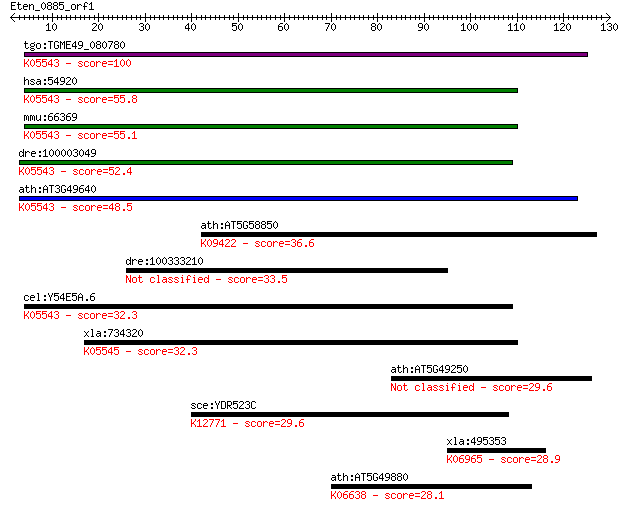
tgo:TGME49_080780 dihydrouridine synthase domain-containing pr... 100 1e-21
hsa:54920 DUS2L, DUS2, FLJ20399, SMM1, URLC8; dihydrouridine s... 55.8 3e-08
mmu:66369 Dus2l, 2310016K04Rik; dihydrouridine synthase 2-like... 55.1 5e-08
dre:100003049 dus2l; dihydrouridine synthase 2-like, SMM1 homo... 52.4 3e-07
ath:AT3G49640 FAD binding / catalytic/ tRNA dihydrouridine syn... 48.5 5e-06
ath:AT5G58850 MYB119; MYB119 (MYB DOMAIN PROTEIN 119); DNA bin... 36.6 0.022
dre:100333210 mannose receptor C type 1-like 33.5 0.15
cel:Y54E5A.6 hypothetical protein; K05543 tRNA-dihydrouridine ... 32.3 0.43
xla:734320 dus4l; dihydrouridine synthase 4-like; K05545 tRNA-... 32.3 0.43
ath:AT5G49250 beta-galactosidase 29.6 2.5
sce:YDR523C SPS1; Putative protein serine/threonine kinase exp... 29.6 2.7
xla:495353 pelo, pelota; pelota homolog; K06965 protein pelota 28.9 3.9
ath:AT5G49880 mitotic checkpoint family protein; K06638 mitoti... 28.1 6.3
> tgo:TGME49_080780 dihydrouridine synthase domain-containing
protein ; K05543 tRNA-dihydrouridine synthase 2 [EC:1.-.-.-]
Length=653
Score = 100 bits (248), Expect = 1e-21, Method: Compositional matrix adjust.
Identities = 62/165 (37%), Positives = 86/165 (52%), Gaps = 44/165 (26%)
Query 4 DATRFQDSYHIGNLMFARGAMWDPSLFAQKKVVT--------TEGTAACSFE-------- 47
DA FQ+ +LMFARGAMWDPS+F+ KK T ++ +AA S E
Sbjct 465 DADVFQEKIGADSLMFARGAMWDPSIFSWKKGTTKNVVAEFDSDSSAALSCEKETPSLYT 524
Query 48 -------------------------RTAVLQDYIKRAVLCGSPYQSIKFTLQEMTAR-DS 81
R V++ YIKRAV+CG+PYQ+IKF LQ M A+ S
Sbjct 525 SDPPPMGQNTDFSHPGSSLPSSESSRVKVMRGYIKRAVVCGAPYQAIKFCLQSMAAQCSS 584
Query 82 DKKLKLDLVAANSTAKLCDVFGLGDFYKSI--EHVPYANTLNYYK 124
D+ + L + ++ ST LC L FY+++ + P+ANTLNY+K
Sbjct 585 DRNVNLAITSSKSTRDLCRALDLDLFYETVFPKLPPHANTLNYHK 629
> hsa:54920 DUS2L, DUS2, FLJ20399, SMM1, URLC8; dihydrouridine
synthase 2-like, SMM1 homolog (S. cerevisiae); K05543 tRNA-dihydrouridine
synthase 2 [EC:1.-.-.-]
Length=493
Score = 55.8 bits (133), Expect = 3e-08, Method: Compositional matrix adjust.
Identities = 33/106 (31%), Positives = 54/106 (50%), Gaps = 10/106 (9%)
Query 4 DATRFQDSYHIGNLMFARGAMWDPSLFAQKKVVTTEGTAACSFERTAVLQDYIKRAVLCG 63
D F+ + ++M AR AMW+PS+F ++ + E V+Q YI+ AV
Sbjct 226 DIEDFRQATAASSVMVARAAMWNPSIFLKEGLRPLE----------EVMQKYIRYAVQYD 275
Query 64 SPYQSIKFTLQEMTARDSDKKLKLDLVAANSTAKLCDVFGLGDFYK 109
+ Y + K+ L +M + L AA S+ ++C+ FGLG FY+
Sbjct 276 NHYTNTKYCLCQMLREQLESPQGRLLHAAQSSREICEAFGLGAFYE 321
> mmu:66369 Dus2l, 2310016K04Rik; dihydrouridine synthase 2-like
(SMM1, S. cerevisiae); K05543 tRNA-dihydrouridine synthase
2 [EC:1.-.-.-]
Length=493
Score = 55.1 bits (131), Expect = 5e-08, Method: Compositional matrix adjust.
Identities = 33/106 (31%), Positives = 53/106 (50%), Gaps = 10/106 (9%)
Query 4 DATRFQDSYHIGNLMFARGAMWDPSLFAQKKVVTTEGTAACSFERTAVLQDYIKRAVLCG 63
D F+ + ++M AR AMW+PS+F + + E V+Q YI+ AV
Sbjct 226 DIEDFRQATAASSVMVARAAMWNPSIFLKDGLRPLE----------EVMQKYIRYAVQYD 275
Query 64 SPYQSIKFTLQEMTARDSDKKLKLDLVAANSTAKLCDVFGLGDFYK 109
+ Y + K+ L +M + L AA S+ ++C+ FGLG FY+
Sbjct 276 NHYTNTKYCLCQMLREQLESPQGRLLHAAQSSQEICEAFGLGAFYE 321
> dre:100003049 dus2l; dihydrouridine synthase 2-like, SMM1 homolog
(S. cerevisiae); K05543 tRNA-dihydrouridine synthase 2
[EC:1.-.-.-]
Length=504
Score = 52.4 bits (124), Expect = 3e-07, Method: Compositional matrix adjust.
Identities = 30/106 (28%), Positives = 54/106 (50%), Gaps = 10/106 (9%)
Query 3 EDATRFQDSYHIGNLMFARGAMWDPSLFAQKKVVTTEGTAACSFERTAVLQDYIKRAVLC 62
ED F++S ++M AR AMW+PS+F Q+ ++ + V+Q+YI AV
Sbjct 230 EDIETFRESTGASSVMLARAAMWNPSVFRQQGALSVD----------QVMQEYITYAVRY 279
Query 63 GSPYQSIKFTLQEMTARDSDKKLKLDLVAANSTAKLCDVFGLGDFY 108
+ + K+ L +M + L AA + ++C+VFG+ + +
Sbjct 280 DNNPFNTKYCLCQMLRDRVESPFGKRLHAAQTNQEICEVFGMTELF 325
> ath:AT3G49640 FAD binding / catalytic/ tRNA dihydrouridine synthase;
K05543 tRNA-dihydrouridine synthase 2 [EC:1.-.-.-]
Length=290
Score = 48.5 bits (114), Expect = 5e-06, Method: Compositional matrix adjust.
Identities = 31/123 (25%), Positives = 66/123 (53%), Gaps = 13/123 (10%)
Query 3 EDATRFQDSYHIGNLMFARGAMWDPSLFAQKKVVTTEGTAACSFERTAVLQDYIKRAVLC 62
+D +R + + ++M ARGAMW+ S+F+ K E V + Y+++++L
Sbjct 176 DDFSRIKTATGAASVMVARGAMWNASIFSPKGKSHWED----------VKKKYLRKSILW 225
Query 63 GSPYQSIKFTLQEMTARDSDKKLK--LDLVAANSTAKLCDVFGLGDFYKSIEHV-PYANT 119
+ +S K+T++EM A S +L + A++ L ++ L D++ +++++ P +
Sbjct 226 NNDVKSTKYTIKEMIAHHSCLELPEGKSINKADTLEDLARLYDLEDYFWTVKNIGPLTHN 285
Query 120 LNY 122
LN+
Sbjct 286 LNH 288
> ath:AT5G58850 MYB119; MYB119 (MYB DOMAIN PROTEIN 119); DNA binding
/ transcription factor; K09422 myb proto-oncogene protein,
plant
Length=430
Score = 36.6 bits (83), Expect = 0.022, Method: Composition-based stats.
Identities = 26/85 (30%), Positives = 43/85 (50%), Gaps = 13/85 (15%)
Query 42 AACSFERTAVLQDYIKRAVLCGSPYQSIKFTLQEMTARDSDKKLKLDLVAANSTAKLCDV 101
A+ S +R +LQDYIK SI+ + +D+D+K + ++ ST L +
Sbjct 224 ASPSAKRPCILQDYIK----------SIE---RNNINKDNDEKKNENTISVISTPNLDQI 270
Query 102 FGLGDFYKSIEHVPYANTLNYYKHI 126
+ GD SI PY L+Y+++I
Sbjct 271 YSDGDSASSILGGPYDEELDYFQNI 295
> dre:100333210 mannose receptor C type 1-like
Length=326
Score = 33.5 bits (75), Expect = 0.15, Method: Compositional matrix adjust.
Identities = 22/75 (29%), Positives = 35/75 (46%), Gaps = 6/75 (8%)
Query 26 DPSLFAQKKVVTTEGTAACSFERTAVLQDYI---KRAVLCGSPYQSIKFTLQEMTARDSD 82
DP+LF +G A C+F R D K +C + + + F QEMT RD+
Sbjct 87 DPALFLNWASGQPDGNADCTFIRNGQWHDLPCTKKCRFICYNMSRGLVFVKQEMTWRDAQ 146
Query 83 KKLK---LDLVAANS 94
+ +DLV+ ++
Sbjct 147 SHCRQNHVDLVSVSN 161
> cel:Y54E5A.6 hypothetical protein; K05543 tRNA-dihydrouridine
synthase 2 [EC:1.-.-.-]
Length=436
Score = 32.3 bits (72), Expect = 0.43, Method: Compositional matrix adjust.
Identities = 27/107 (25%), Positives = 48/107 (44%), Gaps = 13/107 (12%)
Query 4 DATRFQDSYHIGNLMFARGAMWDPSLFAQKKVVTTEGTAACSFERTAVLQDYIKRAVLCG 63
D ++Q + M AR A+ PS+F + EG ++ ++++++ A
Sbjct 220 DFEKYQLLTETSSTMIARKALSTPSIFRR------EGC----LDKYEDIRNFLELACQYD 269
Query 64 SPYQSIKFTLQEMTARDS--DKKLKLDLVAANSTAKLCDVFGLGDFY 108
Y K+ +Q + D D + K VAA S ++C FG+ D Y
Sbjct 270 ESYTMTKYVVQRILGADQEYDPRGKA-TVAAGSVLQICKAFGMEDVY 315
> xla:734320 dus4l; dihydrouridine synthase 4-like; K05545 tRNA-dihydrouridine
synthase 4 [EC:1.-.-.-]
Length=308
Score = 32.3 bits (72), Expect = 0.43, Method: Compositional matrix adjust.
Identities = 26/95 (27%), Positives = 45/95 (47%), Gaps = 17/95 (17%)
Query 17 LMFARGAMWDPSLFAQKKVVTTEGTAACSFERT--AVLQDYIKRAVLCGSPYQSIKFTLQ 74
+M ARG + +P++FA +E T A +QD++ A+ G+P+ + L
Sbjct 227 VMAARGLLANPAMFA-------------GYEETPLACIQDWVDIALEHGTPFTCLHHHLM 273
Query 75 EMTARDSDKKLKLDLVAANSTAKLCDVFGLGDFYK 109
M R + K+ K +ST+ + D L D Y+
Sbjct 274 YMMERITSKQEKRIFNILSSTSAVLDY--LRDHYE 306
> ath:AT5G49250 beta-galactosidase
Length=200
Score = 29.6 bits (65), Expect = 2.5, Method: Compositional matrix adjust.
Identities = 12/43 (27%), Positives = 29/43 (67%), Gaps = 1/43 (2%)
Query 83 KKLKLDLVAANSTAKLCDVFGLGDFYKSIEHVPYANTLNYYKH 125
K+LK+ ++++ + + C + G+G F+ + H+P +++L +KH
Sbjct 52 KELKIVVLSSLNRSYCCGICGIGLFF-FVAHLPLSSSLFVFKH 93
> sce:YDR523C SPS1; Putative protein serine/threonine kinase expressed
at the end of meiosis and localized to the prospore
membrane, required for correct localization of enzymes involved
in spore wall synthesis (EC:2.7.11.1); K12771 sporulation-specific
protein 1 [EC:2.7.11.1]
Length=490
Score = 29.6 bits (65), Expect = 2.7, Method: Compositional matrix adjust.
Identities = 20/72 (27%), Positives = 32/72 (44%), Gaps = 12/72 (16%)
Query 40 GTAACSFERTAVLQDYIKRAVLCGSPYQSIKFTLQEMTA----RDSDKKLKLDLVAANST 95
G +CS D +KR+ + G P + + F + E+T +K+ D+ AAN
Sbjct 97 GGGSCS--------DLLKRSYVNGLPEEKVSFIIHEVTLGLKYLHEQRKIHRDIKAANIL 148
Query 96 AKLCDVFGLGDF 107
+ LGDF
Sbjct 149 LNEEGMVKLGDF 160
> xla:495353 pelo, pelota; pelota homolog; K06965 protein pelota
Length=383
Score = 28.9 bits (63), Expect = 3.9, Method: Compositional matrix adjust.
Identities = 10/21 (47%), Positives = 14/21 (66%), Gaps = 0/21 (0%)
Query 95 TAKLCDVFGLGDFYKSIEHVP 115
T C++ LGDFYK ++H P
Sbjct 269 TKAACEIKALGDFYKMLQHEP 289
> ath:AT5G49880 mitotic checkpoint family protein; K06638 mitotic
spindle assembly checkpoint protein MAD1
Length=726
Score = 28.1 bits (61), Expect = 6.3, Method: Composition-based stats.
Identities = 14/43 (32%), Positives = 24/43 (55%), Gaps = 0/43 (0%)
Query 70 KFTLQEMTARDSDKKLKLDLVAANSTAKLCDVFGLGDFYKSIE 112
+FTLQ + A+ D+KL+ + + N++ + GD K IE
Sbjct 657 RFTLQSIYAQSDDEKLEFEYESGNTSILNNEYASQGDIAKQIE 699
Lambda K H
0.320 0.133 0.395
Gapped
Lambda K H
0.267 0.0410 0.140
Effective search space used: 2044474180
Database: egene_temp_file_orthology_annotation_similarity_blast_database_866
Posted date: Sep 17, 2011 2:57 PM
Number of letters in database: 82,071,388
Number of sequences in database: 164,496
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Neighboring words threshold: 11
Window for multiple hits: 40