bitscore colors: <40, 40-50 , 50-80, 80-200, >200
BLASTP 2.2.24+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs", Nucleic Acids Res. 25:3389-3402.
Reference for composition-based statistics: Alejandro A. Schaffer,
L. Aravind, Thomas L. Madden, Sergei Shavirin, John L. Spouge, Yuri
I. Wolf, Eugene V. Koonin, and Stephen F. Altschul (2001),
"Improving the accuracy of PSI-BLAST protein database searches with
composition-based statistics and other refinements", Nucleic Acids
Res. 29:2994-3005.
Database: egene_temp_file_orthology_annotation_similarity_blast_database_866
164,496 sequences; 82,071,388 total letters
Query= Eten_1223_orf2
Length=160
Score E
Sequences producing significant alignments: (Bits) Value
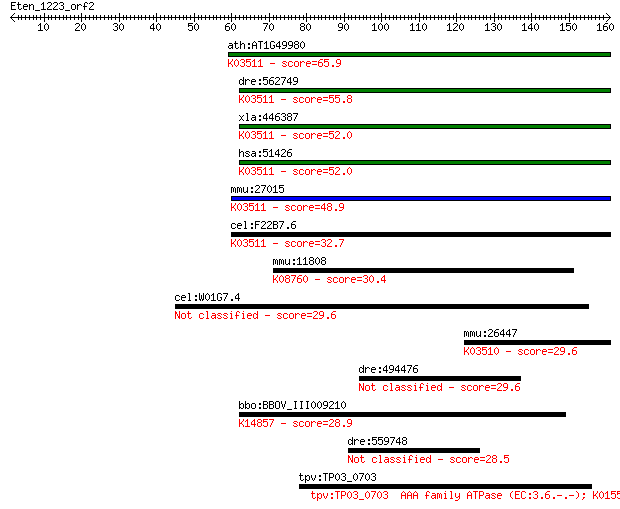
ath:AT1G49980 DNA-directed DNA polymerase/ damaged DNA binding... 65.9 5e-11
dre:562749 polk, si:ch211-254o18.3; polymerase (DNA directed) ... 55.8 7e-08
xla:446387 polk, MGC83722; polymerase (DNA directed) kappa (EC... 52.0 7e-07
hsa:51426 POLK, DINB1, DINP, POLQ; polymerase (DNA directed) k... 52.0 9e-07
mmu:27015 Polk, Dinb1; polymerase (DNA directed), kappa (EC:2.... 48.9 7e-06
cel:F22B7.6 polk-1; POLK (DNA polymerase kappa) homolog family... 32.7 0.52
mmu:11808 Apoa4, Apoa-4; apolipoprotein A-IV; K08760 apolipopr... 30.4 2.4
cel:W01G7.4 hypothetical protein 29.6 4.1
mmu:26447 Poli, Rad30b; polymerase (DNA directed), iota (EC:2.... 29.6 4.6
dre:494476 tfr2, MGC123043, zgc:123043; transferrin receptor 2 29.6
bbo:BBOV_III009210 17.m10612; ribosomal RNA large subunit meth... 28.9 6.7
dre:559748 hypothetical LOC559748 28.5 8.7
tpv:TP03_0703 AAA family ATPase (EC:3.6.-.-); K01554 [EC:3.6.... 28.5 9.7
> ath:AT1G49980 DNA-directed DNA polymerase/ damaged DNA binding;
K03511 DNA polymerase kappa subunit [EC:2.7.7.7]
Length=671
Score = 65.9 bits (159), Expect = 5e-11, Method: Compositional matrix adjust.
Identities = 34/102 (33%), Positives = 60/102 (58%), Gaps = 5/102 (4%)
Query 59 IFANSKAGMDMTEEEKAKIAAKIFELSKNSPFYANEMRKNKQMEQQLSVLRRRVQLFASS 118
+F N+KAGM+ ++EK + ++E+SK S ++ NE RK M+Q++ +R R +S
Sbjct 22 VFTNAKAGMEGVDKEK--VQRVVYEMSKGSKYFQNEERKEALMKQKIEHMRDRCAKLSSL 79
Query 119 GGSAAASATARRVLQQLQQQQQRESGRVYVHLDMDMFFCAVE 160
S +R+ L+ + R+ R+++H+DMD F+ AVE
Sbjct 80 DLSNYQKVVDKRI---LELEATRDLSRIWLHVDMDAFYAAVE 118
> dre:562749 polk, si:ch211-254o18.3; polymerase (DNA directed)
kappa; K03511 DNA polymerase kappa subunit [EC:2.7.7.7]
Length=888
Score = 55.8 bits (133), Expect = 7e-08, Method: Compositional matrix adjust.
Identities = 36/100 (36%), Positives = 59/100 (59%), Gaps = 7/100 (7%)
Query 62 NSKAGMDMTEEEKAKIAAKIFELSKNSPFYANEMRKNKQMEQQLS-VLRRRVQLFASSGG 120
++KAGM+ + +K I I E SK S FY NE++K +Q+ Q++ ++ ++ ++ +
Sbjct 36 DNKAGMEGLDRDK--INKIILETSKGSRFYENELKKERQVNQRIEKMMEQKAKI--TKEQ 91
Query 121 SAAASATARRVLQQLQQQQQRESGRVYVHLDMDMFFCAVE 160
A A R+ L++ RE GRV VH+DMD F+ AVE
Sbjct 92 MKQAQAEVDRLTVDLERS--RELGRVIVHVDMDAFYAAVE 129
> xla:446387 polk, MGC83722; polymerase (DNA directed) kappa (EC:2.7.7.7);
K03511 DNA polymerase kappa subunit [EC:2.7.7.7]
Length=862
Score = 52.0 bits (123), Expect = 7e-07, Method: Compositional matrix adjust.
Identities = 34/100 (34%), Positives = 61/100 (61%), Gaps = 7/100 (7%)
Query 62 NSKAGMDMTEEEKAKIAAKIFELSKNSPFYANEMRKNKQMEQQLS-VLRRRVQLFASSGG 120
++KAGM+ ++EK I I + +K S FY NE++K++Q+ Q++ +++++ QL +S
Sbjct 24 DNKAGMEGLDKEK--INKIIMDATKGSRFYDNELKKDQQVNQRIEKMMQQKSQL--TSQQ 79
Query 121 SAAASATARRVLQQLQQQQQRESGRVYVHLDMDMFFCAVE 160
A R+ L ++ R+ R+ VH+DMD F+ AVE
Sbjct 80 IRKAQIQVDRL--ALALEKNRDLSRIIVHVDMDAFYAAVE 117
> hsa:51426 POLK, DINB1, DINP, POLQ; polymerase (DNA directed)
kappa (EC:2.7.7.7); K03511 DNA polymerase kappa subunit [EC:2.7.7.7]
Length=870
Score = 52.0 bits (123), Expect = 9e-07, Method: Compositional matrix adjust.
Identities = 35/100 (35%), Positives = 58/100 (58%), Gaps = 7/100 (7%)
Query 62 NSKAGMDMTEEEKAKIAAKIFELSKNSPFYANEMRKNKQMEQQL-SVLRRRVQLFASSGG 120
++KAGM+ ++EK I I E +K S FY NE++K KQ+ Q++ ++++++ Q+ +S
Sbjct 23 DNKAGMEGLDKEK--INKIIMEATKGSRFYGNELKKEKQVNQRIENMMQQKAQI--TSQQ 78
Query 121 SAAASATARRVLQQLQQQQQRESGRVYVHLDMDMFFCAVE 160
A R +L +Q R VH+DMD F+ AVE
Sbjct 79 LRKAQLQVDRFAMEL--EQSRNLSNTIVHIDMDAFYAAVE 116
> mmu:27015 Polk, Dinb1; polymerase (DNA directed), kappa (EC:2.7.7.7);
K03511 DNA polymerase kappa subunit [EC:2.7.7.7]
Length=852
Score = 48.9 bits (115), Expect = 7e-06, Method: Compositional matrix adjust.
Identities = 33/102 (32%), Positives = 58/102 (56%), Gaps = 7/102 (6%)
Query 60 FANSKAGMDMTEEEKAKIAAKIFELSKNSPFYANEMRKNKQMEQQL-SVLRRRVQLFASS 118
++KAGM+ ++EK I I E +K S FY NE++K KQ+ Q++ ++++++ Q+ +S
Sbjct 20 LNDNKAGMEGLDKEK--INKIIMEATKGSRFYGNELKKEKQVNQRIENMMQQKAQI--TS 75
Query 119 GGSAAASATARRVLQQLQQQQQRESGRVYVHLDMDMFFCAVE 160
A + +L ++ R VH+DMD F+ AVE
Sbjct 76 QQLRKAQLQVDKFAMEL--ERNRNLNNTIVHVDMDAFYAAVE 115
> cel:F22B7.6 polk-1; POLK (DNA polymerase kappa) homolog family
member (polk-1); K03511 DNA polymerase kappa subunit [EC:2.7.7.7]
Length=596
Score = 32.7 bits (73), Expect = 0.52, Method: Composition-based stats.
Identities = 23/103 (22%), Positives = 54/103 (52%), Gaps = 10/103 (9%)
Query 60 FANSKAGMDMTEEEKAKIAAKIFELSKNSPFYANEMRKNKQMEQQLSVLRRRVQLFASSG 119
F ++KAGM+ ++EK K+ E + ++ + + ++ ++E+++ ++ R+Q
Sbjct 4 FNDNKAGMNGLDKEKI---TKVIEENTSASYSSFSKKQQSRIEEKVLEIKNRLQT----- 55
Query 120 GSAAASATARRVLQQLQQ--QQQRESGRVYVHLDMDMFFCAVE 160
+ + +++ L+ + R+ R V +DMD +F AVE
Sbjct 56 ATREERQKSEILMENLEMKLESSRDLSRDCVCIDMDAYFAAVE 98
> mmu:11808 Apoa4, Apoa-4; apolipoprotein A-IV; K08760 apolipoprotein
A-IV
Length=395
Score = 30.4 bits (67), Expect = 2.4, Method: Compositional matrix adjust.
Identities = 29/96 (30%), Positives = 41/96 (42%), Gaps = 21/96 (21%)
Query 71 EEEKAKIAAKIFELSKN-SPFYANEMRK---------------NKQMEQQLSVLRRRVQL 114
EE + K++AKI +L KN +P + K N+Q+EQQ+ RR V+
Sbjct 250 EELQTKVSAKIDQLQKNLAPLVEDVQSKVKGNTEGLQKSLEDLNRQLEQQVEEFRRTVEP 309
Query 115 FASSGGSAAASATARRVLQQLQQQQQRESGRVYVHL 150
A L+Q +QQ SG V HL
Sbjct 310 MGEMFNKALVQQ-----LEQFRQQLGPNSGEVESHL 340
> cel:W01G7.4 hypothetical protein
Length=251
Score = 29.6 bits (65), Expect = 4.1, Method: Compositional matrix adjust.
Identities = 29/116 (25%), Positives = 51/116 (43%), Gaps = 17/116 (14%)
Query 45 AAGPDLETVCRQTFIFANSKAGMDMTEEE------KAKIAAKIFELSKNSPFYANEMRKN 98
AAG DLE + + GM++ +EE K + + E+ KN
Sbjct 41 AAGNDLELNGARVLDLPTQEHGMELADEENPLGFVKDEAVGLVGEVEKNV---------- 90
Query 99 KQMEQQLSVLRRRVQLFASSGGSAAASATARRVLQQLQQQQQRESGRVYVHLDMDM 154
K+ +Q+S+ R +Q + GG AAA ++ V ++R + V +L+ D+
Sbjct 91 KKPAEQVSLNGRLLQP-VTDGGGAAAGSSDDVVYDYYAIHEKRGNPEVVGNLEQDI 145
> mmu:26447 Poli, Rad30b; polymerase (DNA directed), iota (EC:2.7.7.7);
K03510 DNA polymerase iota subunit [EC:2.7.7.7]
Length=737
Score = 29.6 bits (65), Expect = 4.6, Method: Compositional matrix adjust.
Identities = 16/39 (41%), Positives = 22/39 (56%), Gaps = 1/39 (2%)
Query 122 AAASATARRVLQQLQQQQQRESGRVYVHLDMDMFFCAVE 160
A A+ ++R V Q Q S RV VH+D+D F+ VE
Sbjct 26 AGAAGSSRAVCSQ-GPPTQISSSRVIVHVDLDCFYAQVE 63
> dre:494476 tfr2, MGC123043, zgc:123043; transferrin receptor
2
Length=783
Score = 29.6 bits (65), Expect = 5.0, Method: Composition-based stats.
Identities = 16/43 (37%), Positives = 23/43 (53%), Gaps = 0/43 (0%)
Query 94 EMRKNKQMEQQLSVLRRRVQLFASSGGSAAASATARRVLQQLQ 136
EM K E+++ RRV + GS+ +A AR +LQ LQ
Sbjct 129 EMLKKYLKEERIENTLRRVSRESHPPGSSEGNAIAREILQNLQ 171
> bbo:BBOV_III009210 17.m10612; ribosomal RNA large subunit methyltransferase
J family protein; K14857 AdoMet-dependent rRNA
methyltransferase SPB1 [EC:2.1.1.-]
Length=959
Score = 28.9 bits (63), Expect = 6.7, Method: Compositional matrix adjust.
Identities = 30/91 (32%), Positives = 47/91 (51%), Gaps = 8/91 (8%)
Query 62 NSKAGMDMTEEEKAKIAAKIFELSKNSPF-YANEMRKNKQMEQQL---SVLRRRVQLFAS 117
+S + +T+E + AK++EL KN P E R ++M Q SVL R L S
Sbjct 692 HSNYELPVTKELMKRYRAKLYEL-KNRPIRKVLEARGRRKMRVQRKLKSVLPRVEALQNS 750
Query 118 SGGSAAASATARRVLQQLQQQQQRESGRVYV 148
GSA TA+++L+++Q + +VYV
Sbjct 751 DTGSAK---TAKKLLRKVQNTAANKREKVYV 778
> dre:559748 hypothetical LOC559748
Length=211
Score = 28.5 bits (62), Expect = 8.7, Method: Compositional matrix adjust.
Identities = 13/35 (37%), Positives = 21/35 (60%), Gaps = 0/35 (0%)
Query 91 YANEMRKNKQMEQQLSVLRRRVQLFASSGGSAAAS 125
Y ++ N+Q+E+Q+SV+R R+Q S AS
Sbjct 60 YETQLELNEQLEKQISVVRERLQTLRSDPSDRLAS 94
> tpv:TP03_0703 AAA family ATPase (EC:3.6.-.-); K01554 [EC:3.6.-.-]
Length=547
Score = 28.5 bits (62), Expect = 9.7, Method: Composition-based stats.
Identities = 20/78 (25%), Positives = 38/78 (48%), Gaps = 7/78 (8%)
Query 78 AAKIFELSKNSPFYANEMRKNKQMEQQLSVLRRRVQLFASSGGSAAASATARRVLQQLQQ 137
+ K FEL+K EM K ++++ Q+ +R R + A + R+ L QQ
Sbjct 55 SQKAFELTK-----LQEMTKQQELQMQIEQMRLRQGELGTQ--KAKVESDERKKLLSHQQ 107
Query 138 QQQRESGRVYVHLDMDMF 155
+Q+R + + L+ +M+
Sbjct 108 EQERITAQYKAKLEDEMY 125
Lambda K H
0.314 0.122 0.328
Gapped
Lambda K H
0.267 0.0410 0.140
Effective search space used: 3712313100
Database: egene_temp_file_orthology_annotation_similarity_blast_database_866
Posted date: Sep 17, 2011 2:57 PM
Number of letters in database: 82,071,388
Number of sequences in database: 164,496
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Neighboring words threshold: 11
Window for multiple hits: 40