bitscore colors: <40, 40-50 , 50-80, 80-200, >200
BLASTP 2.2.24+
Reference: Stephen F. Altschul, Thomas L. Madden, Alejandro A.
Schaffer, Jinghui Zhang, Zheng Zhang, Webb Miller, and David J.
Lipman (1997), "Gapped BLAST and PSI-BLAST: a new generation of
protein database search programs", Nucleic Acids Res. 25:3389-3402.
Reference for composition-based statistics: Alejandro A. Schaffer,
L. Aravind, Thomas L. Madden, Sergei Shavirin, John L. Spouge, Yuri
I. Wolf, Eugene V. Koonin, and Stephen F. Altschul (2001),
"Improving the accuracy of PSI-BLAST protein database searches with
composition-based statistics and other refinements", Nucleic Acids
Res. 29:2994-3005.
Database: egene_temp_file_orthology_annotation_similarity_blast_database_866
164,496 sequences; 82,071,388 total letters
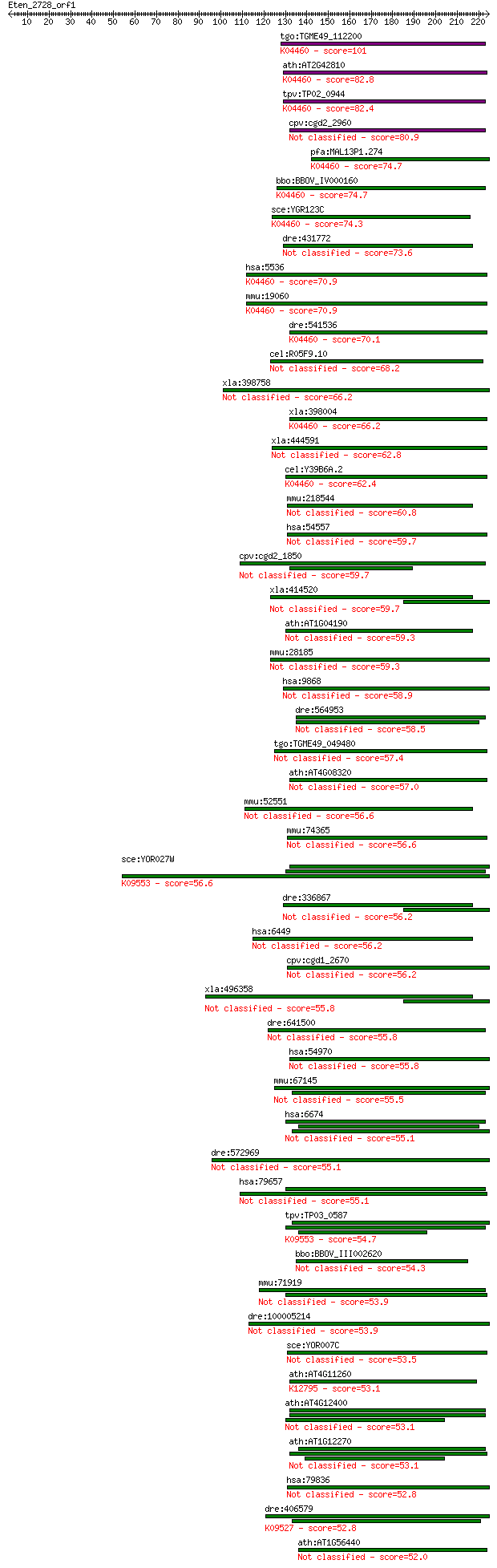
Query= Eten_2728_orf1
Length=224
Score E
Sequences producing significant alignments: (Bits) Value
tgo:TGME49_112200 serine/threonine protein phosphatase, putati... 101 2e-21
ath:AT2G42810 PP5.2; PP5.2 (PROTEIN PHOSPHATASE 5.2); phosphop... 82.8 1e-15
tpv:TP02_0944 serine/threonine protein phosphatase; K04460 pro... 82.4 1e-15
cpv:cgd2_2960 phosphoprotein phosphatase related 80.9 4e-15
pfa:MAL13P1.274 PfPP5; serine/threonine protein phosphatase (E... 74.7 2e-13
bbo:BBOV_IV000160 21.m02802; serine/threonine protein phosphat... 74.7 2e-13
sce:YGR123C PPT1; Ppt1p (EC:3.1.3.16); K04460 protein phosphat... 74.3 3e-13
dre:431772 sgta, zgc:92462; small glutamine-rich tetratricopep... 73.6 5e-13
hsa:5536 PPP5C, FLJ36922, FLJ55954, PP5, PPP5, PPT; protein ph... 70.9 4e-12
mmu:19060 Ppp5c, AU020526, PP5; protein phosphatase 5, catalyt... 70.9 4e-12
dre:541536 fc83f08, im:7146608, ppp5c, wu:fc83f08; zgc:110801 ... 70.1 5e-12
cel:R05F9.10 sgt-1; Small Glutamine-rich Tetratrico repeat pro... 68.2 3e-11
xla:398758 tomm70a, MGC68780; translocase of outer mitochondri... 66.2 8e-11
xla:398004 ppp5c, pp5; protein phosphatase 5, catalytic subuni... 66.2 8e-11
xla:444591 sgtb, MGC84046; small glutamine-rich tetratricopept... 62.8 1e-09
cel:Y39B6A.2 pph-5; Protein PHosphatase family member (pph-5);... 62.4 1e-09
mmu:218544 Sgtb, C630001O05Rik, MGC27660; small glutamine-rich... 60.8 4e-09
hsa:54557 SGTB, FLJ39002, SGT2; small glutamine-rich tetratric... 59.7 7e-09
cpv:cgd2_1850 stress-induced protein sti1-like protein 59.7 8e-09
xla:414520 hypothetical protein MGC81394 59.7 8e-09
ath:AT1G04190 tetratricopeptide repeat (TPR)-containing protein 59.3 9e-09
mmu:28185 Tomm70a, 2610044B22Rik, D16Ium22, D16Ium22e, D16Wsu1... 59.3 1e-08
hsa:9868 TOMM70A, FLJ90470; translocase of outer mitochondrial... 58.9 1e-08
dre:564953 spag1, MGC162178, cb1089, wu:fj78g10; sperm associa... 58.5 2e-08
tgo:TGME49_049480 TPR domain-containing protein 57.4 4e-08
ath:AT4G08320 tetratricopeptide repeat (TPR)-containing protein 57.0 5e-08
mmu:52551 Sgta, 5330427H01Rik, AI194281, D10Ertd190e, MGC6336,... 56.6 6e-08
mmu:74365 Lonrf3, 4932412G04Rik, 5730439E01Rik, A830039N02Rik,... 56.6 8e-08
sce:YOR027W STI1; Hsp90 cochaperone, interacts with the Ssa gr... 56.6 8e-08
dre:336867 fk20d10, wu:fa05b08, wu:fc52b05, wu:fk20d10, zgc:77... 56.2 8e-08
hsa:6449 SGTA, SGT, alphaSGT, hSGT; small glutamine-rich tetra... 56.2 9e-08
cpv:cgd1_2670 ankyrin-related protein 56.2 1e-07
xla:496358 sgta; small glutamine-rich tetratricopeptide repeat... 55.8 1e-07
dre:641500 MGC123010, wu:fk11h08; zgc:123010 55.8 1e-07
hsa:54970 TTC12, FLJ13859, FLJ20535, TPARM; tetratricopeptide ... 55.8 1e-07
mmu:67145 Tomm34, 2610100K07Rik, TOM34, Tomm34a, Tomm34b; tran... 55.5 2e-07
hsa:6674 SPAG1, FLJ32920, HSD-3.8, SP75, TPIS; sperm associate... 55.1 2e-07
dre:572969 tomm70a, KIAA0719, MGC73188, wu:fj58b04, zgc:73188;... 55.1 2e-07
hsa:79657 RPAP3, FLJ21908; RNA polymerase II associated protein 3 55.1
tpv:TP03_0587 hypothetical protein; K09553 stress-induced-phos... 54.7 3e-07
bbo:BBOV_III002620 17.m07250; ankyrin repeat family protein 54.3 3e-07
mmu:71919 Rpap3, 2310042P20Rik, D15Ertd682e; RNA polymerase II... 53.9 4e-07
dre:100005214 TTC3 protein-like 53.9 5e-07
sce:YOR007C SGT2; Glutamine-rich cytoplasmic protein of unknow... 53.5 6e-07
ath:AT4G11260 SGT1B; SGT1B; protein binding; K12795 suppressor... 53.1 7e-07
ath:AT4G12400 stress-inducible protein, putative 53.1 7e-07
ath:AT1G12270 stress-inducible protein, putative 53.1 8e-07
hsa:79836 LONRF3, FLJ22612, MGC119463, MGC119465, RNF127; LON ... 52.8 1e-06
dre:406579 dnajc7, wu:fj58b08, zgc:85806; DnaJ (Hsp40) homolog... 52.8 1e-06
ath:AT1G56440 serine/threonine protein phosphatase-related 52.0 2e-06
> tgo:TGME49_112200 serine/threonine protein phosphatase, putative
(EC:3.1.3.16); K04460 protein phosphatase 5 [EC:3.1.3.16]
Length=548
Score = 101 bits (251), Expect = 2e-21, Method: Compositional matrix adjust.
Identities = 48/96 (50%), Positives = 66/96 (68%), Gaps = 1/96 (1%)
Query 128 VLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQ-LHVFLCNRAFAHL 186
++ AE+LK EGN FK + +AV KY+AAIDL+ + + Q L V LCNRAF +
Sbjct 57 MVAEAESLKTEGNEFFKTRLFHQAVEKYTAAIDLICSNTMTAQTKQILQVLLCNRAFCQI 116
Query 187 RMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
+EN+GSA++DAER +++NP F+K YYRRG Y CL
Sbjct 117 NLENYGSAVVDAERVIQMNPLFAKAYYRRGCAYCCL 152
> ath:AT2G42810 PP5.2; PP5.2 (PROTEIN PHOSPHATASE 5.2); phosphoprotein
phosphatase/ protein binding / protein serine/threonine
phosphatase; K04460 protein phosphatase 5 [EC:3.1.3.16]
Length=538
Score = 82.8 bits (203), Expect = 1e-15, Method: Compositional matrix adjust.
Identities = 39/95 (41%), Positives = 60/95 (63%), Gaps = 9/95 (9%)
Query 129 LERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRM 188
+ RAE K++ N FK H+Y+ A+ Y+ AI+L + V+ NRAFAH ++
Sbjct 10 VSRAEEFKSQANEAFKGHKYSSAIDLYTKAIEL---------NSNNAVYWANRAFAHTKL 60
Query 189 ENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
E +GSAI DA +A++++ ++SKGYYRRG Y +G
Sbjct 61 EEYGSAIQDASKAIEVDSRYSKGYYRRGAAYLAMG 95
> tpv:TP02_0944 serine/threonine protein phosphatase; K04460 protein
phosphatase 5 [EC:3.1.3.16]
Length=548
Score = 82.4 bits (202), Expect = 1e-15, Method: Compositional matrix adjust.
Identities = 44/108 (40%), Positives = 64/108 (59%), Gaps = 14/108 (12%)
Query 129 LERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAP------------VDP--RETQL 174
LE+AE K EGN +F ++ + A+ YS +I LV+++ + P R+T L
Sbjct 54 LEKAELKKLEGNKMFSENNFLSAIEHYSESIRLVEDSHLVSNFKKEGYNWITPELRKTNL 113
Query 175 HVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
H + NRA ++++EN+GSAI DA A++L P F K YYRRG Y CL
Sbjct 114 HQYYSNRAICNIKIENYGSAISDANMAIELRPDFFKAYYRRGCAYLCL 161
> cpv:cgd2_2960 phosphoprotein phosphatase related
Length=525
Score = 80.9 bits (198), Expect = 4e-15, Method: Compositional matrix adjust.
Identities = 41/91 (45%), Positives = 56/91 (61%), Gaps = 1/91 (1%)
Query 132 AEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENF 191
+E K +GN FK +Y EA+ Y+ AI +A + + LH++ NRA H+R+ENF
Sbjct 14 SEQYKIKGNESFKSGKYNEAIEYYTLAIK-TSQASNETQNKNLHIYYSNRALCHIRLENF 72
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
GSAI D+ ++K P FSK YYRRG YF L
Sbjct 73 GSAIEDSGESIKCCPSFSKAYYRRGIAYFNL 103
> pfa:MAL13P1.274 PfPP5; serine/threonine protein phosphatase
(EC:3.1.3.16); K04460 protein phosphatase 5 [EC:3.1.3.16]
Length=658
Score = 74.7 bits (182), Expect = 2e-13, Method: Compositional matrix adjust.
Identities = 32/83 (38%), Positives = 53/83 (63%), Gaps = 8/83 (9%)
Query 142 LFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFGSAIIDAERA 201
LFK++ A++K S I + +ET LH++ NR+F H+++EN+G+AI D + A
Sbjct 200 LFKEYYNKSAISKKSDFISI--------KETDLHIYYTNRSFCHIKLENYGTAIEDIDEA 251
Query 202 LKLNPKFSKGYYRRGTGYFCLGN 224
+K+NP ++K YYR+G Y L +
Sbjct 252 IKINPYYAKAYYRKGCSYLLLSD 274
> bbo:BBOV_IV000160 21.m02802; serine/threonine protein phosphatase
5 (EC:3.1.3.16); K04460 protein phosphatase 5 [EC:3.1.3.16]
Length=545
Score = 74.7 bits (182), Expect = 2e-13, Method: Compositional matrix adjust.
Identities = 42/113 (37%), Positives = 60/113 (53%), Gaps = 16/113 (14%)
Query 126 AAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDL----VDEAPVDPR----------- 170
A +RA+ + EGN F + Y AV Y+ AI + ++EA V
Sbjct 19 AEAQQRADEKRLEGNKFFGEGDYPVAVELYTQAIGILRKAIEEANVRQNSENSTNVDTLS 78
Query 171 -ETQLHVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
+T +H NRA H+RMEN+G A++DA+ A+ P++SK YYRRG Y CL
Sbjct 79 SQTNIHQLYTNRALCHIRMENYGLAVLDADAAIMAQPEYSKAYYRRGCAYICL 131
> sce:YGR123C PPT1; Ppt1p (EC:3.1.3.16); K04460 protein phosphatase
5 [EC:3.1.3.16]
Length=513
Score = 74.3 bits (181), Expect = 3e-13, Method: Compositional matrix adjust.
Identities = 38/94 (40%), Positives = 59/94 (62%), Gaps = 11/94 (11%)
Query 124 ASAAVLERAEAL--KNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNR 181
++ +RA+AL KNEGNV K+ + +A+ KY+ AIDL ++ ++ NR
Sbjct 2 STPTAADRAKALERKNEGNVFVKEKHFLKAIEKYTEAIDL---------DSTQSIYFSNR 52
Query 182 AFAHLRMENFGSAIIDAERALKLNPKFSKGYYRR 215
AFAH +++NF SA+ D + A+KL+PK K Y+RR
Sbjct 53 AFAHFKVDNFQSALNDCDEAIKLDPKNIKAYHRR 86
> dre:431772 sgta, zgc:92462; small glutamine-rich tetratricopeptide
repeat (TPR)-containing, alpha
Length=306
Score = 73.6 bits (179), Expect = 5e-13, Method: Compositional matrix adjust.
Identities = 40/88 (45%), Positives = 54/88 (61%), Gaps = 9/88 (10%)
Query 129 LERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRM 188
+ERAE LKNEGN K+ Y+ AV Y+ AI+L D R V+ CNRA AH ++
Sbjct 84 IERAEQLKNEGNNHMKEENYSSAVDCYTKAIEL------DQRNA---VYYCNRAAAHSKL 134
Query 189 ENFGSAIIDAERALKLNPKFSKGYYRRG 216
EN+ A+ D ERA+ ++P +SK Y R G
Sbjct 135 ENYTEAMGDCERAIAIDPSYSKAYGRMG 162
> hsa:5536 PPP5C, FLJ36922, FLJ55954, PP5, PPP5, PPT; protein
phosphatase 5, catalytic subunit (EC:3.1.3.16); K04460 protein
phosphatase 5 [EC:3.1.3.16]
Length=477
Score = 70.9 bits (172), Expect = 4e-12, Method: Compositional matrix adjust.
Identities = 43/114 (37%), Positives = 61/114 (53%), Gaps = 11/114 (9%)
Query 112 GNVVPCSE--KDVSASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDP 169
G C+E +D + L+RAE LK + N FK Y A+ YS AI+L +P
Sbjct 6 GERTECAEPPRDEPPADGALKRAEELKTQANDYFKAKDYENAIKFYSQAIEL------NP 59
Query 170 RETQLHVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
++ NR+ A+LR E +G A+ DA RA++L+ K+ KGYYRR LG
Sbjct 60 SNA---IYYGNRSLAYLRTECYGYALGDATRAIELDKKYIKGYYRRAASNMALG 110
> mmu:19060 Ppp5c, AU020526, PP5; protein phosphatase 5, catalytic
subunit (EC:3.1.3.16); K04460 protein phosphatase 5 [EC:3.1.3.16]
Length=499
Score = 70.9 bits (172), Expect = 4e-12, Method: Compositional matrix adjust.
Identities = 43/114 (37%), Positives = 61/114 (53%), Gaps = 11/114 (9%)
Query 112 GNVVPCSE--KDVSASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDP 169
G C+E +D + L+RAE LK + N FK Y A+ YS AI+L +P
Sbjct 6 GERTECAETPRDEPPADGTLKRAEELKTQANDYFKAKDYENAIKFYSQAIEL------NP 59
Query 170 RETQLHVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
++ NR+ A+LR E +G A+ DA RA++L+ K+ KGYYRR LG
Sbjct 60 GNA---IYYGNRSLAYLRTECYGYALGDATRAIELDKKYIKGYYRRAASNMALG 110
> dre:541536 fc83f08, im:7146608, ppp5c, wu:fc83f08; zgc:110801
(EC:3.1.3.16); K04460 protein phosphatase 5 [EC:3.1.3.16]
Length=481
Score = 70.1 bits (170), Expect = 5e-12, Method: Compositional matrix adjust.
Identities = 37/92 (40%), Positives = 52/92 (56%), Gaps = 9/92 (9%)
Query 132 AEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENF 191
AE LK + N FK Y A+ Y+ A+DL P +P ++ NR+ ++LR E +
Sbjct 10 AEKLKEKANDYFKDKDYENAIKYYTEALDL---NPTNP------IYYSNRSLSYLRTECY 60
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
G A+ DA RAL+L+ + KGYYRR T LG
Sbjct 61 GYALADATRALELDKNYLKGYYRRATSNMALG 92
> cel:R05F9.10 sgt-1; Small Glutamine-rich Tetratrico repeat protein
family member (sgt-1)
Length=337
Score = 68.2 bits (165), Expect = 3e-11, Method: Compositional matrix adjust.
Identities = 41/99 (41%), Positives = 54/99 (54%), Gaps = 10/99 (10%)
Query 123 SASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRA 182
+ S + + +A LK EGN L K Q+ AV KY+AAI L DP V+ CNRA
Sbjct 96 TPSDSDISQANKLKEEGNDLMKASQFEAAVQKYNAAIKLN----RDP------VYFCNRA 145
Query 183 FAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFC 221
A+ R+E + AI D AL L+P +SK + R G Y C
Sbjct 146 AAYCRLEQYDLAIQDCRTALALDPSYSKAWGRMGLAYSC 184
> xla:398758 tomm70a, MGC68780; translocase of outer mitochondrial
membrane 70 homolog A
Length=576
Score = 66.2 bits (160), Expect = 8e-11, Method: Compositional matrix adjust.
Identities = 40/124 (32%), Positives = 65/124 (52%), Gaps = 7/124 (5%)
Query 101 DGGGSCAAKEAGNVVPCSEKDVSASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAID 160
+G S E G+ P +D + +E+A+A KN+GN FK +Y +A+ Y+ AI
Sbjct 54 EGSASPVPSEGGSNNP---QDAPQELSPIEKAQAAKNKGNKYFKASKYEQAIQCYTEAIS 110
Query 161 LVDEAPVDPRETQLHVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYF 220
L P ++ L F NRA AH + +N+ + D +A++LNP++ K +RR +
Sbjct 111 LC---PAH-NKSDLSTFYQNRAAAHEQSQNWKEVVEDCTKAVELNPRYVKALFRRAKAHE 166
Query 221 CLGN 224
L N
Sbjct 167 KLDN 170
> xla:398004 ppp5c, pp5; protein phosphatase 5, catalytic subunit
(EC:3.1.3.16); K04460 protein phosphatase 5 [EC:3.1.3.16]
Length=493
Score = 66.2 bits (160), Expect = 8e-11, Method: Compositional matrix adjust.
Identities = 36/92 (39%), Positives = 50/92 (54%), Gaps = 9/92 (9%)
Query 132 AEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENF 191
AE LK + N F+ Y AV Y+ AIDL + + + NR+ A+LR E +
Sbjct 22 AEELKEQANEYFRVKDYDHAVQYYTQAIDLSPDTAI---------YYGNRSLAYLRTECY 72
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
G A+ DA RA++L+ K+ KGYYRR LG
Sbjct 73 GYALADASRAIQLDAKYIKGYYRRAASNMALG 104
> xla:444591 sgtb, MGC84046; small glutamine-rich tetratricopeptide
repeat (TPR)-containing, beta
Length=308
Score = 62.8 bits (151), Expect = 1e-09, Method: Compositional matrix adjust.
Identities = 38/100 (38%), Positives = 52/100 (52%), Gaps = 9/100 (9%)
Query 124 ASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAF 183
+S + E+AE LK+EGN L K+ Y AV YS AI+L DP V+ CNRA
Sbjct 81 SSLSAAEKAEQLKDEGNGLMKEQNYEAAVDCYSQAIEL------DPNNA---VYYCNRAA 131
Query 184 AHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
A + AI D E+A+ ++ K+SK Y R G +
Sbjct 132 AQSQRGKHSEAITDCEKAISIDAKYSKAYGRMGRALVAMS 171
> cel:Y39B6A.2 pph-5; Protein PHosphatase family member (pph-5);
K04460 protein phosphatase 5 [EC:3.1.3.16]
Length=496
Score = 62.4 bits (150), Expect = 1e-09, Method: Compositional matrix adjust.
Identities = 36/94 (38%), Positives = 51/94 (54%), Gaps = 10/94 (10%)
Query 130 ERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRME 189
E+A +K+E N FK Y A YS AI++ A V NRA A+L+ E
Sbjct 27 EKAGMIKDEANQFFKDQVYDVAADLYSVAIEIHPTA----------VLYGNRAQAYLKKE 76
Query 190 NFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
+GSA+ DA+ A+ ++P + KG+YRR T LG
Sbjct 77 LYGSALEDADNAIAIDPSYVKGFYRRATANMALG 110
> mmu:218544 Sgtb, C630001O05Rik, MGC27660; small glutamine-rich
tetratricopeptide repeat (TPR)-containing, beta
Length=304
Score = 60.8 bits (146), Expect = 4e-09, Method: Compositional matrix adjust.
Identities = 34/86 (39%), Positives = 50/86 (58%), Gaps = 9/86 (10%)
Query 131 RAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMEN 190
+A+ LK+EGN K+ YA AV Y+ AI+L DP V+ CNRA A ++ +
Sbjct 84 KADQLKDEGNNHMKEENYAAAVDCYTQAIEL------DPNNA---VYYCNRAAAQSKLSH 134
Query 191 FGSAIIDAERALKLNPKFSKGYYRRG 216
+ AI D E+A+ ++ K+SK Y R G
Sbjct 135 YTDAIKDCEKAIAIDSKYSKAYGRMG 160
> hsa:54557 SGTB, FLJ39002, SGT2; small glutamine-rich tetratricopeptide
repeat (TPR)-containing, beta
Length=304
Score = 59.7 bits (143), Expect = 7e-09, Method: Compositional matrix adjust.
Identities = 35/93 (37%), Positives = 51/93 (54%), Gaps = 9/93 (9%)
Query 131 RAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMEN 190
+A+ LK+EGN K+ YA AV Y+ AI+L DP V+ CNRA A ++ +
Sbjct 84 KADQLKDEGNNHMKEENYAAAVDCYTQAIEL------DPNNA---VYYCNRAAAQSKLGH 134
Query 191 FGSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
+ AI D E+A+ ++ K+SK Y R G L
Sbjct 135 YTDAIKDCEKAIAIDSKYSKAYGRMGLALTALN 167
> cpv:cgd2_1850 stress-induced protein sti1-like protein
Length=326
Score = 59.7 bits (143), Expect = 8e-09, Method: Compositional matrix adjust.
Identities = 38/114 (33%), Positives = 64/114 (56%), Gaps = 12/114 (10%)
Query 109 KEAGNVVPCSEKDVSASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVD 168
KE ++ +EK+ + E AE + EGN LFKQ Y A +Y AI +
Sbjct 120 KELERLIEKAEKEAYINP---ELAEKHRIEGNDLFKQKNYPAAKKEYDEAI------KRN 170
Query 169 PRETQLHVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
P +++L+ NRA ++++ + SA+ID ++AL L+PKF+K + R+G ++ L
Sbjct 171 PSDSRLY---SNRAACYMQLLEYPSALIDVQKALDLDPKFTKAWSRKGNIHYFL 221
Score = 38.1 bits (87), Expect = 0.027, Method: Compositional matrix adjust.
Identities = 21/57 (36%), Positives = 32/57 (56%), Gaps = 9/57 (15%)
Query 132 AEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRM 188
AE KN+GN L+KQ ++ EA+ +Y AI+ +DP + FL N+ +L M
Sbjct 5 AEFYKNKGNELYKQKKFDEALVQYDLAIE------IDPNDIS---FLTNKGAVYLEM 52
> xla:414520 hypothetical protein MGC81394
Length=312
Score = 59.7 bits (143), Expect = 8e-09, Method: Compositional matrix adjust.
Identities = 34/94 (36%), Positives = 51/94 (54%), Gaps = 9/94 (9%)
Query 123 SASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRA 182
S S + AE+LK EGN K + AV Y+ A++L +PR V+ CNRA
Sbjct 79 SPSDEDVAEAESLKTEGNEQMKVENFESAVTYYTKALEL------NPRNA---VYYCNRA 129
Query 183 FAHLRMENFGSAIIDAERALKLNPKFSKGYYRRG 216
A+ ++ N+ A+ D E A+ ++P +SK Y R G
Sbjct 130 AAYSKLGNYAGAVRDCEEAISIDPSYSKAYGRMG 163
Score = 34.7 bits (78), Expect = 0.27, Method: Compositional matrix adjust.
Identities = 16/40 (40%), Positives = 24/40 (60%), Gaps = 0/40 (0%)
Query 185 HLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
+++ENF SA+ +AL+LNP+ + Y R Y LGN
Sbjct 98 QMKVENFESAVTYYTKALELNPRNAVYYCNRAAAYSKLGN 137
> ath:AT1G04190 tetratricopeptide repeat (TPR)-containing protein
Length=328
Score = 59.3 bits (142), Expect = 9e-09, Method: Compositional matrix adjust.
Identities = 34/87 (39%), Positives = 48/87 (55%), Gaps = 9/87 (10%)
Query 130 ERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRME 189
E ++LK +GN FK + +A A Y+ AI L DP L+ NRA A L +
Sbjct 13 EAEKSLKEKGNEFFKAGNFLKAAALYTQAIKL------DPSNATLY---SNRAAAFLSLV 63
Query 190 NFGSAIIDAERALKLNPKFSKGYYRRG 216
A+ DAE +KLNP++ KGY+R+G
Sbjct 64 KLSKALADAETTIKLNPQWEKGYFRKG 90
> mmu:28185 Tomm70a, 2610044B22Rik, D16Ium22, D16Ium22e, D16Wsu109e,
Tom70, mKIAA0719; translocase of outer mitochondrial
membrane 70 homolog A (yeast)
Length=611
Score = 59.3 bits (142), Expect = 1e-08, Method: Compositional matrix adjust.
Identities = 35/102 (34%), Positives = 55/102 (53%), Gaps = 4/102 (3%)
Query 123 SASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRA 182
S + L+RA+A KN+GN FK +Y +A+ Y+ AI L P + + L F NRA
Sbjct 108 SLEMSSLDRAQAAKNKGNKYFKAGKYEQAIQCYTEAISLC---PTE-KNVDLSTFYQNRA 163
Query 183 FAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
A +++ + D +A++LNPK+ K +RR + L N
Sbjct 164 AAFEQLQKWKEVAQDCTKAVELNPKYVKALFRRAKAHEKLDN 205
> hsa:9868 TOMM70A, FLJ90470; translocase of outer mitochondrial
membrane 70 homolog A (S. cerevisiae)
Length=608
Score = 58.9 bits (141), Expect = 1e-08, Method: Compositional matrix adjust.
Identities = 34/96 (35%), Positives = 53/96 (55%), Gaps = 4/96 (4%)
Query 129 LERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRM 188
L+RA+A KN+GN FK +Y +A+ Y+ AI L P + + L F NRA A ++
Sbjct 111 LDRAQAAKNKGNKYFKAGKYEQAIQCYTEAISLC---PTE-KNVDLSTFYQNRAAAFEQL 166
Query 189 ENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
+ + D +A++LNPK+ K +RR + L N
Sbjct 167 QKWKEVAQDCTKAVELNPKYVKALFRRAKAHEKLDN 202
> dre:564953 spag1, MGC162178, cb1089, wu:fj78g10; sperm associated
antigen 1
Length=386
Score = 58.5 bits (140), Expect = 2e-08, Method: Compositional matrix adjust.
Identities = 38/88 (43%), Positives = 48/88 (54%), Gaps = 1/88 (1%)
Query 135 LKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFGSA 194
LKN+GN+LFK Q+ +A+ KY+ AID EA +D E L V NRA L+ N
Sbjct 87 LKNQGNMLFKNGQFGDALEKYTQAIDGCIEAGIDSPED-LCVLYSNRAACFLKDGNSADC 145
Query 195 IIDAERALKLNPKFSKGYYRRGTGYFCL 222
I D RAL+L+P K RR Y L
Sbjct 146 IQDCTRALELHPFSLKPLLRRAMAYESL 173
Score = 50.4 bits (119), Expect = 5e-06, Method: Compositional matrix adjust.
Identities = 30/85 (35%), Positives = 42/85 (49%), Gaps = 9/85 (10%)
Query 135 LKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFGSA 194
LK EGN L K Q+ A KYS + + P E ++ NRA L++E F A
Sbjct 264 LKQEGNELVKNSQFQGASEKYSECL------AIKPNECAIYT---NRALCFLKLERFAEA 314
Query 195 IIDAERALKLNPKFSKGYYRRGTGY 219
D + AL++ PK K +YRR +
Sbjct 315 KQDCDSALQMEPKNKKAFYRRALAH 339
> tgo:TGME49_049480 TPR domain-containing protein
Length=1161
Score = 57.4 bits (137), Expect = 4e-08, Method: Compositional matrix adjust.
Identities = 40/131 (30%), Positives = 57/131 (43%), Gaps = 32/131 (24%)
Query 125 SAAVLERAEALKNEGNVLFKQHQYAEAVAKYS----AAIDLVDEAP-------------- 166
S A+L R +ALK EGN FK+ ++ A+ YS A D +D+ P
Sbjct 5 SNAMLARLQALKEEGNAEFKRGKFESAIEAYSRCLDDASDTLDKEPDVLGGACAASLSSS 64
Query 167 ----VDPRETQLHVF----------LCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGY 212
+PR+ + LCNRA + R + F +A D RA+ L+P + K Y
Sbjct 65 DSQVAEPRKESPAILKRVAELKAQILCNRALCYQRTKQFAAAEADCTRAIALHPAYVKSY 124
Query 213 YRRGTGYFCLG 223
YRR G
Sbjct 125 YRRAVALDAQG 135
> ath:AT4G08320 tetratricopeptide repeat (TPR)-containing protein
Length=427
Score = 57.0 bits (136), Expect = 5e-08, Method: Compositional matrix adjust.
Identities = 34/92 (36%), Positives = 49/92 (53%), Gaps = 9/92 (9%)
Query 132 AEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENF 191
AE LK +GN + + Y EAV YS AI L D+ VF CNRA A+ ++
Sbjct 175 AETLKCQGNKAMQSNLYLEAVELYSFAIALTDKN---------AVFYCNRAAAYTQINMC 225
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
AI D ++++++P +SK Y R G Y+ G
Sbjct 226 SEAIKDCLKSIEIDPNYSKAYSRLGLAYYAQG 257
> mmu:52551 Sgta, 5330427H01Rik, AI194281, D10Ertd190e, MGC6336,
Sgt, Stg; small glutamine-rich tetratricopeptide repeat (TPR)-containing,
alpha
Length=315
Score = 56.6 bits (135), Expect = 6e-08, Method: Compositional matrix adjust.
Identities = 38/106 (35%), Positives = 54/106 (50%), Gaps = 16/106 (15%)
Query 111 AGNVVPCSEKDVSASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPR 170
A + P SE+D + AE LK EGN K + AV Y AI+L +P
Sbjct 78 APDRTPPSEEDSA-------EAERLKTEGNEQMKLENFEAAVHLYGKAIEL------NPA 124
Query 171 ETQLHVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRG 216
V+ CNRA A+ ++ N+ A+ D ERA+ ++P +SK Y R G
Sbjct 125 NA---VYFCNRAAAYSKLGNYVGAVQDCERAIGIDPGYSKAYGRMG 167
> mmu:74365 Lonrf3, 4932412G04Rik, 5730439E01Rik, A830039N02Rik,
AU023707, Rnf127; LON peptidase N-terminal domain and ring
finger 3
Length=753
Score = 56.6 bits (135), Expect = 8e-08, Method: Compositional matrix adjust.
Identities = 34/93 (36%), Positives = 50/93 (53%), Gaps = 9/93 (9%)
Query 131 RAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMEN 190
RA L++EGN LF++HQ A+ KY+ A+ L AP D H+ NR+ + +E+
Sbjct 243 RASQLRHEGNRLFREHQVEAALLKYNEAVRL---APND------HLLYSNRSQIYFTLES 293
Query 191 FGSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
A+ DAE A KL P K ++R+ LG
Sbjct 294 HEDALHDAEIACKLRPMGFKAHFRKAQALATLG 326
> sce:YOR027W STI1; Hsp90 cochaperone, interacts with the Ssa
group of the cytosolic Hsp70 chaperones; activates the ATPase
activity of Ssa1p; homolog of mammalian Hop protein; K09553
stress-induced-phosphoprotein 1
Length=589
Score = 56.6 bits (135), Expect = 8e-08, Method: Compositional matrix adjust.
Identities = 30/93 (32%), Positives = 48/93 (51%), Gaps = 8/93 (8%)
Query 132 AEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENF 191
A+ K +GN F Y +A+ ++ AI++ ET HV NR+ + ++ F
Sbjct 5 ADEYKQQGNAAFTAKDYDKAIELFTKAIEVS--------ETPNHVLYSNRSACYTSLKKF 56
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
A+ DA +K+NP +SKGY R G + LG+
Sbjct 57 SDALNDANECVKINPSWSKGYNRLGAAHLGLGD 89
Score = 43.1 bits (100), Expect = 7e-04, Method: Compositional matrix adjust.
Identities = 27/93 (29%), Positives = 45/93 (48%), Gaps = 9/93 (9%)
Query 130 ERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRME 189
E+AE + EG F + + AV Y+ +++ AP D R NRA A ++
Sbjct 394 EKAEEARLEGKEYFTKSDWPNAVKAYT---EMIKRAPEDAR------GYSNRAAALAKLM 444
Query 190 NFGSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
+F AI D +A++ +P F + Y R+ T +
Sbjct 445 SFPEAIADCNKAIEKDPNFVRAYIRKATAQIAV 477
Score = 37.4 bits (85), Expect = 0.039, Method: Compositional matrix adjust.
Identities = 46/188 (24%), Positives = 77/188 (40%), Gaps = 31/188 (16%)
Query 54 IFGVTQLLNRRLCLMAEGSDSAAISCNSAESLKREEGQIIKDSAMAVDG---GGSCAAKE 110
+F +L+ LM G D N + S+ +E + K + D S +KE
Sbjct 178 LFTDPRLMTIMATLM--GVDLNMDDINQSNSMPKE-PETSKSTEQKKDAEPQSDSTTSKE 234
Query 111 AGNVVPCSEK-------DVSASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVD 163
+ P E+ +V + +E A+ K EGN +K Q+ EA+ Y+ A +L
Sbjct 235 NSSKAPQKEESKESEPMEVDEDDSKIE-ADKEKAEGNKFYKARQFDEAIEHYNKAWELHK 293
Query 164 EAPVDPRETQLHVFLCNRAFAHLRMENFGSAI------IDAERALKLNPK-FSKGYYRRG 216
+ +L NRA A + +AI ++ R ++ + K SK + R G
Sbjct 294 DI----------TYLNNRAAAEYEKGEYETAISTLNDAVEQGREMRADYKVISKSFARIG 343
Query 217 TGYFCLGN 224
Y LG+
Sbjct 344 NAYHKLGD 351
> dre:336867 fk20d10, wu:fa05b08, wu:fc52b05, wu:fk20d10, zgc:77080;
zgc:55741
Length=320
Score = 56.2 bits (134), Expect = 8e-08, Method: Compositional matrix adjust.
Identities = 33/88 (37%), Positives = 46/88 (52%), Gaps = 9/88 (10%)
Query 129 LERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRM 188
L AE LK +GN K ++ AV YS AI L Q V+ CNRA A+ ++
Sbjct 88 LAEAERLKTDGNDQMKVENFSAAVEFYSKAIQL---------NPQNAVYFCNRAAAYSKL 138
Query 189 ENFGSAIIDAERALKLNPKFSKGYYRRG 216
N+ A+ D ERA+ ++ +SK Y R G
Sbjct 139 GNYAGAVQDCERAIGIDANYSKAYGRMG 166
Score = 30.4 bits (67), Expect = 5.6, Method: Compositional matrix adjust.
Identities = 13/40 (32%), Positives = 24/40 (60%), Gaps = 0/40 (0%)
Query 185 HLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
+++ENF +A+ +A++LNP+ + + R Y LGN
Sbjct 101 QMKVENFSAAVEFYSKAIQLNPQNAVYFCNRAAAYSKLGN 140
> hsa:6449 SGTA, SGT, alphaSGT, hSGT; small glutamine-rich tetratricopeptide
repeat (TPR)-containing, alpha
Length=313
Score = 56.2 bits (134), Expect = 9e-08, Method: Compositional matrix adjust.
Identities = 37/102 (36%), Positives = 52/102 (50%), Gaps = 16/102 (15%)
Query 115 VPCSEKDVSASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQL 174
P SE+D + AE LK EGN K + AV Y AI+L +P
Sbjct 81 TPPSEEDSA-------EAERLKTEGNEQMKVENFEAAVHFYGKAIEL------NPANA-- 125
Query 175 HVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRG 216
V+ CNRA A+ ++ N+ A+ D ERA+ ++P +SK Y R G
Sbjct 126 -VYFCNRAAAYSKLGNYAGAVQDCERAICIDPAYSKAYGRMG 166
> cpv:cgd1_2670 ankyrin-related protein
Length=503
Score = 56.2 bits (134), Expect = 1e-07, Method: Compositional matrix adjust.
Identities = 30/97 (30%), Positives = 53/97 (54%), Gaps = 3/97 (3%)
Query 131 RAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHV---FLCNRAFAHLR 187
AE LK E N FK+ Q+ +++ YS A+ + + E + + L NR+ ++++
Sbjct 367 EAERLKAEANSFFKEGQFEKSIEMYSNALSHLMMKKNNLSEGAITLKSSILSNRSLSYIK 426
Query 188 MENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
+ + A+ DA + +NPK+SKGYYR Y +G+
Sbjct 427 IGDSSKALFDATSCIYINPKWSKGYYRCSQVYHMIGD 463
> xla:496358 sgta; small glutamine-rich tetratricopeptide repeat
(TPR)-containing, alpha
Length=302
Score = 55.8 bits (133), Expect = 1e-07, Method: Compositional matrix adjust.
Identities = 41/126 (32%), Positives = 62/126 (49%), Gaps = 11/126 (8%)
Query 93 IKDSAMAVDGGGSCAAKEAG--NVVPCSEKDVSASAAVLERAEALKNEGNVLFKQHQYAE 150
I+DS++AV EA N + S S L AE LK EGN K +
Sbjct 35 IEDSSLAVPQTLQEIFTEATFQNSPEVNSGLASPSDEDLAEAERLKTEGNEQMKVENFES 94
Query 151 AVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSK 210
A++ Y+ A++L +P V+ CNRA A+ ++ N+ A+ D E A+ ++P +SK
Sbjct 95 AISYYTKALEL------NPANA---VYYCNRAAAYSKLGNYAGAVRDCEAAITIDPNYSK 145
Query 211 GYYRRG 216
Y R G
Sbjct 146 AYGRMG 151
Score = 33.1 bits (74), Expect = 0.79, Method: Compositional matrix adjust.
Identities = 17/40 (42%), Positives = 23/40 (57%), Gaps = 0/40 (0%)
Query 185 HLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
+++ENF SAI +AL+LNP + Y R Y LGN
Sbjct 86 QMKVENFESAISYYTKALELNPANAVYYCNRAAAYSKLGN 125
> dre:641500 MGC123010, wu:fk11h08; zgc:123010
Length=474
Score = 55.8 bits (133), Expect = 1e-07, Method: Compositional matrix adjust.
Identities = 30/101 (29%), Positives = 57/101 (56%), Gaps = 9/101 (8%)
Query 122 VSASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNR 181
+ S A +R+ +L +G ++ QY +AV+ ++ AI DP++ + F NR
Sbjct 174 IGFSEAKTKRSASLVEKGIRFVQEGQYTQAVSLFTEAIK------CDPKD---YRFFGNR 224
Query 182 AFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
++ + +E + A+ DAE+++++ P + KGYYRRG+ L
Sbjct 225 SYCYCCLEQYALALADAEKSIQMAPDWPKGYYRRGSALMGL 265
> hsa:54970 TTC12, FLJ13859, FLJ20535, TPARM; tetratricopeptide
repeat domain 12
Length=705
Score = 55.8 bits (133), Expect = 1e-07, Method: Compositional matrix adjust.
Identities = 29/93 (31%), Positives = 50/93 (53%), Gaps = 9/93 (9%)
Query 132 AEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENF 191
A+ALK +GN F + Y A+ +YS ++ + + + V NRA A++++E++
Sbjct 106 ADALKEKGNEAFAEGNYETAILRYSEGLEKLKD---------MKVLYTNRAQAYMKLEDY 156
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
A++D E ALK + K +K Y+ G L N
Sbjct 157 EKALVDCEWALKCDEKCTKAYFHMGKANLALKN 189
> mmu:67145 Tomm34, 2610100K07Rik, TOM34, Tomm34a, Tomm34b; translocase
of outer mitochondrial membrane 34
Length=309
Score = 55.5 bits (132), Expect = 2e-07, Method: Compositional matrix adjust.
Identities = 36/102 (35%), Positives = 53/102 (51%), Gaps = 13/102 (12%)
Query 125 SAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAI--DLVDEAPVDPRETQLHVFLCNRA 182
SA +ERA+ALK EGN L K+ + +A+ KYS ++ ++ A NRA
Sbjct 186 SAGDVERAKALKEEGNDLVKKGNHKKAIEKYSESLLCSSLESA-----------TYSNRA 234
Query 183 FAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
HL ++ + A+ D ALKL+ K K +YRR Y L +
Sbjct 235 LCHLVLKQYKEAVKDCTEALKLDGKNVKAFYRRAQAYKALKD 276
Score = 42.4 bits (98), Expect = 0.001, Method: Compositional matrix adjust.
Identities = 34/91 (37%), Positives = 41/91 (45%), Gaps = 3/91 (3%)
Query 133 EALKNEGNVLFKQHQYAEAVAKYSAAIDLVD-EAPVDPRETQLHVFLCNRAFAHLRMENF 191
E L+ GN F+ QYAEA A Y A+ L+ DP E V NRA +L+ N
Sbjct 10 EELRAAGNQSFRNGQYAEASALYERALRLLQARGSADPEEE--SVLYSNRAACYLKDGNC 67
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
I D AL L P K RR + Y L
Sbjct 68 TDCIKDCTSALALVPFSIKPLLRRASAYEAL 98
> hsa:6674 SPAG1, FLJ32920, HSD-3.8, SP75, TPIS; sperm associated
antigen 1
Length=926
Score = 55.1 bits (131), Expect = 2e-07, Method: Compositional matrix adjust.
Identities = 37/93 (39%), Positives = 50/93 (53%), Gaps = 1/93 (1%)
Query 130 ERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRME 189
E LK++GN LF+ Q+AEA KYSAAI L++ A + + L + NRA +L+
Sbjct 443 ENPAGLKSQGNELFRSGQFAEAAGKYSAAIALLEPAGSEIAD-DLSILYSNRAACYLKEG 501
Query 190 NFGSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
N I D RAL+L+P K RR Y L
Sbjct 502 NCSGCIQDCNRALELHPFSMKPLLRRAMAYETL 534
Score = 44.7 bits (104), Expect = 3e-04, Method: Compositional matrix adjust.
Identities = 28/84 (33%), Positives = 39/84 (46%), Gaps = 10/84 (11%)
Query 136 KNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFGSAI 195
K +GN F Y EAV Y+ +I + V NRA A ++++N+ SA
Sbjct 213 KEKGNEAFNSGDYEEAVMYYTRSISALPTV----------VAYNNRAQAEIKLQNWNSAF 262
Query 196 IDAERALKLNPKFSKGYYRRGTGY 219
D E+ L+L P K RR T Y
Sbjct 263 QDCEKVLELEPGNVKALLRRATTY 286
Score = 40.0 bits (92), Expect = 0.006, Method: Compositional matrix adjust.
Identities = 28/92 (30%), Positives = 46/92 (50%), Gaps = 9/92 (9%)
Query 133 EALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFG 192
+ALK EGN Y +A++KYS + ++ +E ++ NRA +L++ F
Sbjct 624 KALKEEGNQCVNDKNYKDALSKYSECLK------INNKECAIYT---NRALCYLKLCQFE 674
Query 193 SAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
A D ++AL+L K +YRR + L N
Sbjct 675 EAKQDCDQALQLADGNVKAFYRRALAHKGLKN 706
> dre:572969 tomm70a, KIAA0719, MGC73188, wu:fj58b04, zgc:73188;
translocase of outer mitochondrial membrane 70 homolog A
(yeast)
Length=578
Score = 55.1 bits (131), Expect = 2e-07, Method: Compositional matrix adjust.
Identities = 41/131 (31%), Positives = 60/131 (45%), Gaps = 20/131 (15%)
Query 96 SAMAVDGGGSCAAKEAGNVVPCSEKDVSASAAVLERAEALKNEGNVLFKQHQYAEAVAKY 155
SA V G E N+ P L+RA++ KN+GN FK +Y A+ Y
Sbjct 60 SASPVQGQHGATNPELENLSP------------LDRAQSAKNKGNKYFKAGKYDHAIKCY 107
Query 156 SAAIDLVDEAPVDPRETQ--LHVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYY 213
+ AI L P+E + L F NRA A+ + + I D +A++LNP++ K +
Sbjct 108 TEAIGLC------PKEKKGDLSTFYQNRAAAYEQQMKWTEVIQDCSQAVELNPRYVKALF 161
Query 214 RRGTGYFCLGN 224
RR L N
Sbjct 162 RRAKALEKLDN 172
> hsa:79657 RPAP3, FLJ21908; RNA polymerase II associated protein
3
Length=631
Score = 55.1 bits (131), Expect = 2e-07, Method: Compositional matrix adjust.
Identities = 34/93 (36%), Positives = 48/93 (51%), Gaps = 9/93 (9%)
Query 130 ERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRME 189
++A LK +GN FKQ +Y EA+ Y+ +D P +P V NRA A+ R++
Sbjct 131 QKALVLKEKGNKYFKQGKYDEAIDCYTKGMDA---DPYNP------VLPTNRASAYFRLK 181
Query 190 NFGSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
F A D A+ LN ++K Y RRG F L
Sbjct 182 KFAVAESDCNLAVALNRSYTKAYSRRGAARFAL 214
Score = 43.5 bits (101), Expect = 6e-04, Method: Compositional matrix adjust.
Identities = 35/117 (29%), Positives = 57/117 (48%), Gaps = 11/117 (9%)
Query 109 KEAGNVVPCSEKDVSASAAVLERAEAL--KNEGNVLFKQHQYAEAVAKYSAAIDLVDEAP 166
KEA V+ +E + A + +A+ K+ GN FK+ +Y A+ Y+ I D A
Sbjct 257 KEADIVIKSTEGERKQIEAQQNKQQAISEKDRGNGFFKEGKYERAIECYTRGI-AADGAN 315
Query 167 VDPRETQLHVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
+ NRA A+L+++ + A D +A+ L+ +SK + RRGT LG
Sbjct 316 A--------LLPANRAMAYLKIQKYEEAEKDCTQAILLDGSYSKAFARRGTARTFLG 364
> tpv:TP03_0587 hypothetical protein; K09553 stress-induced-phosphoprotein
1
Length=540
Score = 54.7 bits (130), Expect = 3e-07, Method: Compositional matrix adjust.
Identities = 34/92 (36%), Positives = 49/92 (53%), Gaps = 9/92 (9%)
Query 133 EALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFG 192
E LKN GN FK ++ +AV ++ AI+L P D HV NR+ A+ M +
Sbjct 2 EDLKNLGNDAFKAGRFMDAVEFFTKAIEL---NPDD------HVLYSNRSGAYASMYMYN 52
Query 193 SAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
A+ DA + + L P + KGY R+G + LGN
Sbjct 53 EALADANKCIDLKPDWPKGYSRKGLCEYKLGN 84
Score = 47.4 bits (111), Expect = 5e-05, Method: Compositional matrix adjust.
Identities = 31/93 (33%), Positives = 50/93 (53%), Gaps = 9/93 (9%)
Query 130 ERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRME 189
E AE + +GN FK ++ EA +Y AI +P + +L+ NRA A L++
Sbjct 353 ELAEQHREKGNEYFKAFKFPEAKKEYDEAI------KRNPTDAKLY---SNRAAALLKLC 403
Query 190 NFGSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
+ SA+ D +AL+L+P F K + R+G + L
Sbjct 404 EYPSALADCNKALELDPTFVKAWARKGNLHVLL 436
Score = 31.6 bits (70), Expect = 2.6, Method: Compositional matrix adjust.
Identities = 20/60 (33%), Positives = 30/60 (50%), Gaps = 9/60 (15%)
Query 136 KNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFGSAI 195
K EGN +KQ ++ EA+ Y+ AI+L DP L N+A +L M ++ I
Sbjct 224 KEEGNNFYKQKKFTEALEMYNKAIEL------DPNNLLLE---NNKAAVYLEMGDYEKCI 274
> bbo:BBOV_III002620 17.m07250; ankyrin repeat family protein
Length=480
Score = 54.3 bits (129), Expect = 3e-07, Method: Compositional matrix adjust.
Identities = 31/80 (38%), Positives = 41/80 (51%), Gaps = 3/80 (3%)
Query 135 LKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFGSA 194
L+ EG VL YA+A A Y+ I L+ DP L +F NR+ +L + A
Sbjct 347 LREEGRVLVANKDYAQACAIYTKGISLL-SGDTDPE--TLSIFYSNRSHTYLMTGDMDKA 403
Query 195 IIDAERALKLNPKFSKGYYR 214
DAE + LNPK+ KGY R
Sbjct 404 KSDAEMCISLNPKWPKGYLR 423
> mmu:71919 Rpap3, 2310042P20Rik, D15Ertd682e; RNA polymerase
II associated protein 3
Length=660
Score = 53.9 bits (128), Expect = 4e-07, Method: Compositional matrix adjust.
Identities = 36/105 (34%), Positives = 51/105 (48%), Gaps = 9/105 (8%)
Query 118 SEKDVSASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVF 177
SE D ++A LK +GN FKQ +Y EA+ Y+ +D P +P V
Sbjct 120 SESDEDGIRVDSQKALVLKEKGNKYFKQGKYDEAIECYTKGMDA---DPYNP------VL 170
Query 178 LCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
NRA A+ R++ F A D A+ L+ ++K Y RRG F L
Sbjct 171 PTNRASAYFRLKKFAVAESDCNLAIALSRTYTKAYARRGAARFAL 215
Score = 42.0 bits (97), Expect = 0.002, Method: Compositional matrix adjust.
Identities = 29/94 (30%), Positives = 48/94 (51%), Gaps = 9/94 (9%)
Query 130 ERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRME 189
++A A K+ GN FK+ +Y +A+ Y+ I D + NRA A+L+++
Sbjct 282 QKAIAEKDLGNGFFKEGKYEQAIECYTRGI-AADRTNA--------LLPANRAMAYLKIQ 332
Query 190 NFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
+ A D +A+ L+ +SK + RRGT LG
Sbjct 333 RYEEAERDCTQAIVLDGSYSKAFARRGTARTFLG 366
> dre:100005214 TTC3 protein-like
Length=715
Score = 53.9 bits (128), Expect = 5e-07, Method: Compositional matrix adjust.
Identities = 36/118 (30%), Positives = 55/118 (46%), Gaps = 15/118 (12%)
Query 113 NVVPCSEKD------VSASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAP 166
VV CS+K + L +++ +KN+GN F++ +Y A+ YS AI
Sbjct 187 QVVSCSKKKALMEMKFEPDSWSLSKSDEMKNKGNEHFQKKKYDVALKWYSKAIKY----- 241
Query 167 VDPRETQLHVFLCNRAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
H+ NRA LR + A+ D +RA+ L P ++KG+YR F LG
Sbjct 242 ----HPNNHILYGNRALCLLRSGKYLKALGDGKRAIVLQPDWAKGHYRFCDALFYLGE 295
> sce:YOR007C SGT2; Glutamine-rich cytoplasmic protein of unknown
function; contains tetratricopeptide (TPR) repeats, which
often mediate protein-protein interactions; has similarity
to human SGT, which is a cochaperone that negatively regulates
Hsp70
Length=346
Score = 53.5 bits (127), Expect = 6e-07, Method: Compositional matrix adjust.
Identities = 29/93 (31%), Positives = 48/93 (51%), Gaps = 9/93 (9%)
Query 131 RAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMEN 190
+AE LK +GN Y A+ KY+ AI ++ P + ++ NRA AH ++
Sbjct 101 KAEDLKMQGNKAMANKDYELAINKYTEAIKVL---PTN------AIYYANRAAAHSSLKE 151
Query 191 FGSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
+ A+ DAE A+ ++P + +GY R G + G
Sbjct 152 YDQAVKDAESAISIDPSYFRGYSRLGFAKYAQG 184
> ath:AT4G11260 SGT1B; SGT1B; protein binding; K12795 suppressor
of G2 allele of SKP1
Length=358
Score = 53.1 bits (126), Expect = 7e-07, Method: Compositional matrix adjust.
Identities = 30/87 (34%), Positives = 46/87 (52%), Gaps = 9/87 (10%)
Query 132 AEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENF 191
A+ L + F + AV YS AIDL DP F +RA A+++++NF
Sbjct 2 AKELAEKAKEAFLDDDFDVAVDLYSKAIDL------DP---NCAAFFADRAQANIKIDNF 52
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTG 218
A++DA +A++L P +K Y R+GT
Sbjct 53 TEAVVDANKAIELEPTLAKAYLRKGTA 79
> ath:AT4G12400 stress-inducible protein, putative
Length=558
Score = 53.1 bits (126), Expect = 7e-07, Method: Compositional matrix adjust.
Identities = 28/91 (30%), Positives = 49/91 (53%), Gaps = 9/91 (9%)
Query 132 AEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENF 191
AE K++GN F YA A+ ++ AI+L +P + H+ NR+ ++ + +
Sbjct 2 AEEAKSKGNAAFSSGDYATAITHFTEAINL---SPTN------HILYSNRSASYASLHRY 52
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
A+ DA++ ++L P +SKGY R G + L
Sbjct 53 EEALSDAKKTIELKPDWSKGYSRLGAAFIGL 83
Score = 52.4 bits (124), Expect = 1e-06, Method: Compositional matrix adjust.
Identities = 32/91 (35%), Positives = 47/91 (51%), Gaps = 9/91 (9%)
Query 132 AEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENF 191
AE + +GN FK+ +Y EAV YS AI P D R NRA + ++
Sbjct 369 AEEEREKGNGFFKEQKYPEAVKHYSEAIK---RNPNDVR------AYSNRAACYTKLGAL 419
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTGYFCL 222
+ DAE+ ++L+P F+KGY R+G F +
Sbjct 420 PEGLKDAEKCIELDPSFTKGYSRKGAIQFFM 450
Score = 38.5 bits (88), Expect = 0.021, Method: Compositional matrix adjust.
Identities = 24/74 (32%), Positives = 38/74 (51%), Gaps = 9/74 (12%)
Query 130 ERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRME 189
E+A K EGNV +K+ + AV Y+ A++L DE +L NRA +L M
Sbjct 228 EKALKEKGEGNVAYKKKDFGRAVEHYTKAMELDDEDI---------SYLTNRAAVYLEMG 278
Query 190 NFGSAIIDAERALK 203
+ I D ++A++
Sbjct 279 KYEECIEDCDKAVE 292
> ath:AT1G12270 stress-inducible protein, putative
Length=572
Score = 53.1 bits (126), Expect = 8e-07, Method: Compositional matrix adjust.
Identities = 28/87 (32%), Positives = 44/87 (50%), Gaps = 9/87 (10%)
Query 136 KNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFGSAI 195
+ +GN FK+ +Y EA+ Y+ AI R H NRA ++ ++ +
Sbjct 387 REKGNDFFKEQKYPEAIKHYTEAIK---------RNPNDHKAYSNRAASYTKLGAMPEGL 437
Query 196 IDAERALKLNPKFSKGYYRRGTGYFCL 222
DAE+ ++L+P FSKGY R+ F L
Sbjct 438 KDAEKCIELDPTFSKGYSRKAAVQFFL 464
Score = 52.0 bits (123), Expect = 2e-06, Method: Compositional matrix adjust.
Identities = 30/92 (32%), Positives = 44/92 (47%), Gaps = 9/92 (9%)
Query 132 AEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENF 191
AE K +GN F + A+ ++ AI L AP + HV NR+ AH + +
Sbjct 2 AEEAKAKGNAAFSSGDFTTAINHFTEAIAL---APTN------HVLFSNRSAAHASLHQY 52
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTGYFCLG 223
A+ DA+ +KL P + KGY R G + L
Sbjct 53 AEALSDAKETIKLKPYWPKGYSRLGAAHLGLN 84
Score = 33.1 bits (74), Expect = 0.90, Method: Compositional matrix adjust.
Identities = 19/65 (29%), Positives = 31/65 (47%), Gaps = 9/65 (13%)
Query 139 GNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFGSAIIDA 198
GN +K+ + A+ YS AI++ DE +L NRA +L M + I D
Sbjct 251 GNAAYKKKDFETAIQHYSTAIEIDDED---------ISYLTNRAAVYLEMGKYNECIEDC 301
Query 199 ERALK 203
+A++
Sbjct 302 NKAVE 306
> hsa:79836 LONRF3, FLJ22612, MGC119463, MGC119465, RNF127; LON
peptidase N-terminal domain and ring finger 3
Length=759
Score = 52.8 bits (125), Expect = 1e-06, Method: Compositional matrix adjust.
Identities = 32/94 (34%), Positives = 50/94 (53%), Gaps = 9/94 (9%)
Query 131 RAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMEN 190
RA L++EGN L+++ Q A+ KY+ A+ L AP D H+ NR+ + +E+
Sbjct 242 RASQLRHEGNRLYRERQVEAALLKYNEAVKL---APND------HLLYSNRSQIYFTLES 292
Query 191 FGSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
+A+ DAE A KL P K ++R+ LG
Sbjct 293 HENALHDAEIACKLRPMGFKAHFRKAQALATLGK 326
> dre:406579 dnajc7, wu:fj58b08, zgc:85806; DnaJ (Hsp40) homolog,
subfamily C, member 7; K09527 DnaJ homolog subfamily C member
7
Length=472
Score = 52.8 bits (125), Expect = 1e-06, Method: Compositional matrix adjust.
Identities = 31/104 (29%), Positives = 53/104 (50%), Gaps = 9/104 (8%)
Query 121 DVSASAAVLERAEALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCN 180
D+++ + AE K +GN + + YAEA Y+ AIDL P+ + N
Sbjct 2 DLTSDEELEREAEGFKEQGNAYYVKKDYAEAFNFYTKAIDLC------PKNAS---YYGN 52
Query 181 RAFAHLRMENFGSAIIDAERALKLNPKFSKGYYRRGTGYFCLGN 224
RA + + + A+ D+++A++L+ F KG+ R G + LGN
Sbjct 53 RAATLMMLSRYREALEDSQQAVRLDDSFMKGHMREGKCHLLLGN 96
Score = 47.0 bits (110), Expect = 6e-05, Method: Compositional matrix adjust.
Identities = 30/89 (33%), Positives = 43/89 (48%), Gaps = 7/89 (7%)
Query 133 EALKNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFL-CNRAFAHLRMENF 191
+A K EGN FK+ Y EA Y+ A+ +DP + + L CNRA ++
Sbjct 242 KAKKEEGNKAFKEGSYEEAYELYTEAL------TIDPNNIKTNAKLYCNRATVGSKLNKL 295
Query 192 GSAIIDAERALKLNPKFSKGYYRRGTGYF 220
AI D +A+KL+ + K Y RR Y
Sbjct 296 EQAIEDCTKAIKLDETYIKAYLRRAQCYM 324
> ath:AT1G56440 serine/threonine protein phosphatase-related
Length=476
Score = 52.0 bits (123), Expect = 2e-06, Method: Compositional matrix adjust.
Identities = 31/88 (35%), Positives = 45/88 (51%), Gaps = 10/88 (11%)
Query 136 KNEGNVLFKQHQYAEAVAKYSAAIDLVDEAPVDPRETQLHVFLCNRAFAHLRMENFGSAI 195
K +GN FKQ ++ EA+ YS +I L A V NRA A+L+++ + A
Sbjct 88 KEQGNEFFKQKKFNEAIDCYSRSIALSPNA----------VTYANRAMAYLKIKRYREAE 137
Query 196 IDAERALKLNPKFSKGYYRRGTGYFCLG 223
+D AL L+ ++ K Y RR T LG
Sbjct 138 VDCTEALNLDDRYIKAYSRRATARKELG 165
Lambda K H
0.321 0.135 0.405
Gapped
Lambda K H
0.267 0.0410 0.140
Effective search space used: 7459475120
Database: egene_temp_file_orthology_annotation_similarity_blast_database_866
Posted date: Sep 17, 2011 2:57 PM
Number of letters in database: 82,071,388
Number of sequences in database: 164,496
Matrix: BLOSUM62
Gap Penalties: Existence: 11, Extension: 1
Neighboring words threshold: 11
Window for multiple hits: 40