Plenary
Lectures
Proteomics
of Eimeria : a focus on host cell invasion
Liz Bromley and
Fiona Tomley
Institute for Animal Health, Compton, Berkshire, RG20
7NN, UK
Apicomplexan parasites
are serious pathogens of man and domestic livestock
for which there is an urgent need to develop novel,
sustainable therapies based on new drugs or vaccines.
The genomes of many species have been sequenced, or
are nearing completion, including those of Plasmodium
falciparum, Cryptosporidium parvum, Theileria parva,
Theileria annulata, Toxoplasma gondii and Eimeria
tenella. Detailed analysis of these genomes will be
invaluable for developing an understanding of the
biology and biochemistry of the Apicomplexa and for
guiding the selection of novel, effective targets
for drug and vaccine design. However, whilst genome
sequences give good indications of the genes that
are present within each parasite, they provide no
clues about the expression or localisation of gene
products, for example whether they are expressed at
all, targeted to specific sub-cellular location, switched
on or off at particular points during the parasite
lifecycles or whether their level of expression is
modulated by certain conditions. It is precisely these
types of questions that proteomics, defined as the
study of the full protein content of an organism,
or an organelle, is designed to answer.
For obligate
intracellular parasites such as the Apicomplexa, the
molecular interactions between parasite and host-cell
surfaces define uniquely each host-parasite relationship.
The essential nature of host cell recognition, attachment
and invasion, the repeated rounds of invasion that
occur during the course of infections and the accessibility
of the extracellular parasite make these interactions
priority targets for intervention. The process of
invasion is more or less conserved between most apicomplexans
and consists broadly of (1) contact of parasite with
host cell (2) reorientation of the parasite to make
apical contact (3) tight attachment of the parasite
apex to the host cell plasma membrane (PM) (4) rapid
invasion of the parasite accompanied by deformation
of the host PM to form a parasitophorous vacuole (5)
pinching off of the PV from the PM. The process is
driven by a parasite actinomyosin contractile motor
and because host cell surface proteins are excluded
from the PV membrane, parasites within the PV remain
isolated from the host cell endocytic pathway. In
recent years, a working hypothesis of apicomplexan
invasion has evolved in which four classes of parasite
proteins, in addition to the motor proteins, are implicated.
These are GPI-linked surface antigens and a variety
of soluble and membrane-bound proteins derived from
the microneme (MIC), rhoptry (ROP) and dense granule
(GRA) secretory organelles. However, as with the process
of invasion itself there are many paradoxes that remain
to be explored concerning apicomplexan secretory organelles.
Chief amongst these is the fact that the number and
type of organelles varies enormously between parasites,
and between different developmental stages of the
same parasite, and may even be absent. Thus micronemes
range from a handful (Plasmodium merozoites and sporozoites)
through to many hundred (Eimeria merozoites) per cell,
rhoptries from zero (Plasmodium ookinetes) or one
(Cryptosporidium sporozoites) to around a dozen (Eimeria
sporozoites) and dense granules from possibly zero
(Eimeria sporozoites) to around a dozen or more (Toxoplasma
tachyzoites).
We have focused attention in our laboratory on defining
the proteomes of the microneme and rhoptry organelles
of Eimeria tenella with a view to understanding the
precise function of these organelles. Using sequences
of 36 characterised microneme, rhoptry and dense granule
proteins from T. gondii and 21 from Plasmodium spp.,
we initially screened E. tenella genome and EST databases
in silico to look for orthologues and homologues.
Whilst we found some degree of conservation between
the microneme proteins of the three species, largely
due to the possession of common adhesive domains,
there was little evidence for conservation of either
rhoptry or dense granule proteins (for example, see
Table below).
Comparison of organelle
proteins between T. gondii and E. tenella
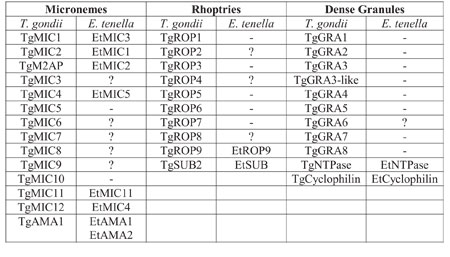
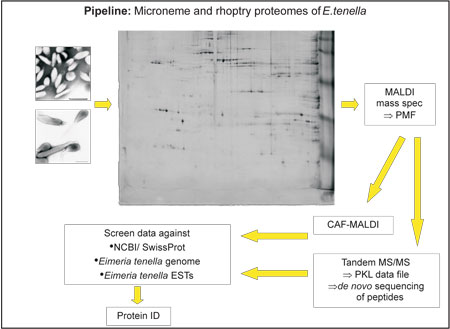
Using proteomics
tools we have now characterised the protein content
of gradient-purified preparations of micronemes and
rhoptries from E.tenella sporozoites. The methods
for purifying these organelles is long-established
in our laboratory (Kawazoe et al, 1992, Parasitology
104, 1-9) and whilst there is some debate about whether
dense granules are present in these stages of Eimeria,
these organelles would most likely be isolated in
the same fraction as the rhoptries. Organellar proteins
were separated by two dimensional gel electrophoresis,
which fractionates proteins on the basis of both their
pI and molecular mass. After staining with a modified
silver stain, protein spots were excised, digested
with trypsin and analysed by various methods including
matrix-assisted laser desorption/ionisation time-of-flight
(MALDI-TOF) mass spectrometry (MS), and tandem electrospray
MS. Interpretation of the mass spectra and comparison
of these data with in silico protein predictions derived
from the Eimeria genome and other databases using
Mascot software resulted in identification of a large
proportion of the protein spots.
Whilst 2D gels
have the advantage over 1D gels of allowing differentiation
between several proteins of the same molecular mass,
they are acknowledged to be unsuitable for the separation
of certain types of protein, including very large
or low abundance proteins, hydrophobic proteins and
proteins of extreme pI. Each large format 2D gel was
loaded with parasite organelle protein purified from
5-10 infected chickens. However even when the technique
was fully optimised with help from 2D gel experts
it was clear that the amounts of proteins visible
on the 2D gels did not reflect the quantities of protein
loaded, particularly for the rhoptry fractions. Therefore
we also separated batches rhoptry proteins by 1D SDS
PAGE and found that many more of the larger proteins
were separated so these were also analysed by Tandem
MS.
The proteins identified
using these approaches fell into three major groups.
In the first group were exogenous non-Eimeria contaminants,
such as trypsin inhibitor, that were introduced during
subcellular fractionation. Second were Eimeria proteins
that from their homologies to other known proteins
are not expected to localise to the apical organelles
and were most likely structural contaminants. The
third group consisted of Eimeria proteins that most
likely encode novel organellar proteins. This final
group included 8 putative novel microneme proteins
and 28 putative novel rhoptry proteins. We have selected
a number of these novel proteins for further study,
as demonstrated by some examples outlined below.
ROPF is a protein
which shares some limited homology to hypothetical
proteins from other apicomplexan parasites so may
represent a novel class of organellar proteins from
these parasites. ROPF is an 200kDa protein excised
from a SDS PAGE gel and the sequences of tryptic peptides
from this band all matched to a single contiguous
genome sequence and to hypothetical proteins from
two species of Plasmodium. Interestingly, peptides
from proteins migrating above and below ROPF on SDS
PAGE in the range 180-220kDa were also found to hit
the same region of the genome Due to the overlap of
the peptides from different proteins along the length
of the gene we have concluded that these proteins
are differently modified forms of expressed gene product.
Sequencing the full-length cDNA of ROPF has revealed
that it occupies >9kb of the genome and is organised
over 22 exons. We have found evidence of extensive
alternative splicing, which accounts for several of
the isoforms and are currently sequencing clones from
a ROPF mini-cDNA library to determine the exact sequence
differences between the isoforms.
ROPJ is a putative
rhoptry protein that shares homology with another
set of Plasmodium hypothetical proteins, as well as
scoring hits onto the genome and/ or ESTs of T. gondii,
N. caninum and T. annulata, suggesting that this protein
belongs to a well-conserved family of rhoptry proteins
from across the whole apicomplexan phylum. We have
sequenced the full-length cDNA of ROPJ, revealing
that the gene is spread over 5kb of the genome, over
12 exons one of which is alternatively spliced between
different cDNAs. ROPJ contains multiple predicted
transmembrane domains and a putative C terminal tyrosine-based
rhoptry sorting signal.
EtSUB is a subtlisin,
a type of serine protease named after its similarity
to a protease which was found to be secreted by the
bacterium Bacillus subtilis. The active site of subtilisins
consists of a characteristic catalytic triad of residues:
aspartic acid, histidine and serine. The catalytic
region of EtSUB has homology with T. gondii subtilisin
TgSUB2, which itself is homologous to the P. falciparum
subtilisin PfSUB2. The apicomplexan subtilisins are
of interest to us as they are variously localised
to all three apical organelles: the microneme (TgSUB1),
the rhoptry (TgSUB2) and the dense granules (PfSUB1
and PfSUB2). Whilst EtSUB appears to be most similar
to the rhoptry enzyme, TgSUB2 we have not yet definitively
localised the enzyme to these organelles. We have
sequenced the full length cDNA of EtSUB, which has
revealed a transcript of approximately 1.1kb encoding
an enzyme of ~114kDa. Again the gene is complex, being
organised over 16 exons with one region of alternative
splicing.
These three
examples of putative rhoptry proteins highlight some
of the characteristic features of E. tenella genes.
They are typically complex, being organised over multiple
exons and employing alternative splicing as a mechanism
for gene expression (see http://www.tigr.org/tigr-scripts/tgi/splnotes.pl?species=e_tenella
for other examples of alternative splicing).
In conclusion,
many novel proteins of E. tenella have been identified
through proteomic analyses of subcellular fractions
of parasites enriched for either microneme or rhoptry
secretory organelles. The relatively complex nature
of these organelles is readily apparent and the analysis
of organelle-specific proteomes of one parasite can
lead to the identification of previously unknown homologues
in other parasites. It is clear that an understanding
of the molecular structure, processing, cellular context
and precise interactions of target molecules will
be critical to the rational development of effective
intervention strategies against apicomplexan parasites.
Also, significant insights into the molecular evolution
of this complex and important group of pathogens can
be obtained by comparative analysis of their organelle-specific
proteomes. In collaboration with experts on Plasmodium
and Toxoplasma we are now expanding work in this area
with the intention of (i) using our expertise in cellular
fractionation to purify organelles from other parasites
where organelles are less abundant (ii) generating
organellar proteomes from important apicomplexan parasites
to build a comprehensive picture of the evolution,
diversity and function of secretory organelles across
the phylum and (iii) exploiting the more tractable
reverse-genetic systems of Toxoplasma gondii and Plasmodium
berghei to begin functional analysis of organellar
proteins that are conserved between different genera,
focusing initially on the genes that we have already
identified in the current work. We are also currently
undertaking a much more comprehensive, high-throughput
proteomics study in which we aim to define whole cell
proteomes for several different developmental stages
of E. tenella using both 2DGE and liquid-chromatography
separations, a process that will not only help to
build up knowledge regarding stage-specific gene regulation
in this parasite but will also served to help and
support the mammoth task of accurate and comprehensive
genome annotation.
We would
like to thank the large number of people who have
contributed to this work including Rich Oakes, Pierre
Rivailler and others from the parasitology group at
IAH; Mike Dunn and colleagues at Proteome Sciences,
London; Jonathan Wastling and colleagues from University
of Liverpool, Bob Sinden and colleagues from Imperial
College and by no means least the entire Eimeria genome
consortium for providing the essential sequence data
that underpins the work.
